Download Biochemistry usmle step 1 and more Cheat Sheet Biochemistry in PDF only on Docsity!
BIOCHEMISTRY ` BIOCHEMISTRY—NUTRITION SEC TION II
Williams syndrome Congenital microdeletion of long arm of chromosome 7 (deleted region includes elastin gene).
Findings: distinctive “elfin” facies, intellectual disability, hypercalcemia, well-developed verbal
skills, extreme friendliness with strangers, cardiovascular problems (eg, supravalvular aortic
stenosis, renal artery stenosis).
` BIOCHEMISTRY—NUTRITION
Essential fatty acids Polyunsaturated fatty acids that cannot be
synthesized in the body and must be provided
in the diet (eg, nuts/seeds, plant oils, seafood).
Linoleic acid (omega-6) is metabolized to
arachidonic acid, which serves as the precursor
to leukotrienes and prostaglandins.
Linolenic acid (omega-3) and its metabolites
have cardioprotective and antihyperlipidemic
effects.
In contrast, consumption of trans-unsaturated
fatty acids (found in fast food) promotes
cardiovascular disease by LDL and HDL.
Vitamins: fat soluble A, D, E, K. Absorption dependent on bile
emulsification, pancreatic secretions, and
intact ileum. Toxicity more common than
for water-soluble vitamins because fat-soluble
vitamins accumulate in fat.
Malabsorption syndromes with steatorrhea (eg,
cystic fibrosis and celiac disease) or mineral
oil intake can cause fat-soluble vitamin
deficiencies.
Vitamins: water
soluble
B
1
(thiamine: TPP)
B
2
(riboflavin: FAD, FMN)
B
3
(niacin: NAD
B
5
(pantothenic acid: CoA)
B
6
(pyridoxine: PLP)
B
7
(biotin)
B
9
(folate)
B
12
(cobalamin)
C (ascorbic acid)
Wash out easily from body except B 12
and B 9
B
12
stored in liver for ~ 3–4 years. B 9
stored in
liver for ~ 3–4 months.
B-complex deficiencies often result in
dermatitis, glossitis, and diarrhea.
Can be coenzymes (eg, ascorbic acid) or
precursors to coenzymes (eg, FAD, NAD
Dietary
supplementation
DIET SUPPLEMENTATION REQUIRED
Vegetarian/vegan Vitamin B
12
Iron
Vitamin B
2
Frequently, vitamin D (although this is commonly deficient in many diets)
High egg white (raw) Vitamin B 7
(avidin in egg whites binds biotin and prevents absorption)
Untreated corn Vitamin B
3
(deficiency is common in resource-limited areas)
SEC TION II BIOCHEMISTRY ` BIOCHEMISTRY—NUTRITION
Vitamin A Includes retinal, retinol, retinoic acid.
FUNCTION Antioxidant; constituent of visual pigments
( retina l); essential for normal differentiation
of epithelial cells into specialized tissue
(pancreatic cells, mucus-secreting cells);
prevents squamous metaplasia.
Retin ol is vitamin A , so think retin-A (used
topically for wrinkles and A cne).
Found in liver and leafy vegetables.
Supplementation in vitamin A-deficient measles
patients may improve outcomes.
Use oral isotretinoin to treat severe cystic acne.
Use all-trans retinoic acid to treat acute
promyelocytic leukemia.
DEFICIENCY
A
Night blindness (nyctalopia); dry, scaly skin
(xerosis cutis); dry eyes (xerophthalmia);
conjunctival squamous metaplasia Bitot
spots (keratin debris; foamy appearance
on conjunctiva A ); corneal degeneration
(keratomalacia); immunosuppression.
EXCESS Acute toxicity—nausea, vomiting, ICP (eg,
vertigo, blurred vision).
Chronic toxicity—alopecia, dry skin (eg,
scaliness), hepatic toxicity and enlargement,
arthralgias, and idiopathic intracranial
hypertension.
Teratogenic (interferes with homeobox gene; cleft
palate, cardiac abnormalities), therefore a
⊝
pregnancy test and two forms of contraception
are required before isotretinoin (vitamin A
derivative) is prescribed.
Is o tret inoin is terat ogenic.
Vitamin B 1
Also called thiamine.
FUNCTION In thiamine pyrophosphate (TPP), a cofactor for several dehydrogenase enzyme reactions ( B e APT ):
B ranched-chain ketoacid dehydrogenase
α-Ketoglutarate dehydrogenase (TCA cycle)
P yruvate dehydrogenase (links glycolysis to TCA cycle)
T ransketolase (HMP shunt)
DEFICIENCY Impaired glucose breakdown ATP depletion worsened by glucose infusion; highly aerobic tissues
(eg, brain, heart) are affected first. In patients with chronic alcohol overuse or malnutrition, give
thiamine before dextrose to risk of precipitating Wernicke encephalopathy.
Diagnosis made by in RBC transketolase activity following vitamin B 1
administration.
DISORDER CHARACTERISTICS
Wernicke
encephalopathy
Acute, reversible, life-threatening neurologic condition. Symptoms: C onfusion, O phthalmoplegia/
N ystagmus, A taxia ( C or ONA beer).
Korsakoff syndrome Amnestic disorder due to chronic alcohol overuse; presents with confabulation, personality
changes, memory loss (permanent).
Wernicke-Korsakoff
syndrome
Damage to medial dorsal nucleus of thalamus, mammillary bodies. Presentation is combination of
Wernicke encephalopathy and Korsakoff syndrome.
Dry beriberi Polyneuropathy, symmetric muscle wasting. Spell beriberi as B er 1B er 1 to remember
vitamin B 1
Wet beriberi High-output cardiac failure (due to systemic
vasodilation).
SEC TION II BIOCHEMISTRY ` BIOCHEMISTRY—NUTRITION
Vitamin B 7
Also called biotin.
FUNCTION Cofactor for carboxylation enzymes (which add a 1-carbon group):
Pyruvate carboxylase (gluconeogenesis): pyruvate (3C)
oxaloacetate (4C)
Acetyl-CoA carboxylase (fatty acid synthesis): acetyl-CoA (2C) malonyl-CoA (3C)
Propionyl-CoA carboxylase (fatty acid oxidation and branched-chain amino acid breakdown):
propionyl-CoA (3C) methylmalonyl-CoA (4C)
DEFICIENCY Relatively rare. Dermatitis, enteritis, alopecia. Caused by long-term antibiotic use or excessive
ingestion of raw egg whites.
“ Avid in in egg whites avid ly binds biotin.”
Vitamin B
9
Also called folate.
FUNCTION Converted to tetrahydrofolic acid (THF), a
coenzyme for 1-carbon transfer/methylation
reactions.
Important for the synthesis of nitrogenous bases
in DNA and RNA.
Found in leafy green vegetables. Also produced
by gut microbiota. Fol ate absorbed in jejun um
(think fol iage in the “ jejun ”gle).
Small reserve pool stored primarily in the liver.
DEFICIENCY Macrocytic, megaloblastic anemia;
hypersegmented polymorphonuclear cells
(PMNs); glossitis; no neurologic symptoms (as
opposed to vitamin B 12
deficiency).
Labs: homocysteine, normal methylmalonic
acid levels. Seen in chronic alcohol overuse
and in pregnancy.
Deficiency can be caused by several drugs (eg,
phenytoin, trimethoprim, methotrexate).
Supplemental folic acid at least 1 month prior
to conception and during pregnancy to risk
of neural tube defects. Give vitamin B
9
for the
9 months of pregnancy, and 1 month prior to
conception.
BIOCHEMISTRY ` BIOCHEMISTRY—NUTRITION SEC TION II
Vitamin B 12
Also called cobalamin.
FUNCTION Cofactor for methionine synthase (transfers
CH
3
groups as methylcobalamin) and
methylmalonyl-CoA mutase. Important for
DNA synthesis.
Found in animal products. Synthesized only
by intestinal microbiota. Site of synthesis in
humans is distal to site of absorption; thus B 12
must be consumed via animal products.
Very large reserve pool (several years) stored
primarily in the liver. Deficiency caused
by malabsorption (eg, sprue, enteritis,
Diphyllobothrium latum, achlorhydria,
bacterial overgrowth, alcohol overuse), lack of
intrinsic factor (eg, pernicious anemia, gastric
bypass surgery), absence of terminal ileum
(surgical resection, eg, for Crohn disease),
certain drugs (eg, metformin), or insufficient
intake (eg, veganism).
B
9
(folate) supplementation can mask the
hematologic symptoms of B
12
deficiency, but
not the neurologic symptoms.
DEFICIENCY Macrocytic, megaloblastic anemia;
hypersegmented PMNs; paresthesias
and subacute combined degeneration
(degeneration of dorsal columns, lateral
corticospinal tracts, and spinocerebellar tracts)
due to abnormal myelin. Associated with
serum homocysteine and methylmalonic
acid levels, along with 2° folate deficiency.
Prolonged deficiency irreversible nerve
damage.
Vitamin C Also called ascorbic acid.
FUNCTION Antioxidant; also facilitates iron absorption
by reducing it to Fe
2+
state. Necessary
for hydroxylation of proline and lysine in
collagen synthesis. Necessary for dopamine
β-hydroxylase (converts dopamine to NE).
Found in fruits and vegetables.
Pronounce “ absorb ic” acid.
Ancillary treatment for methemoglobinemia by
reducing Fe
3+
to Fe
2+
DEFICIENCY Scurvy—swollen gums, easy bruising,
petechiae, hemarthrosis, anemia, poor wound
healing, perifollicular and subperiosteal
hemorrhages, “corkscrew” hair.
Weakened immune response.
Deficiency may be precipitated by tea and toast
diet.
Vitamin C deficiency causes s C urvy due to a
C ollagen hydro C ylation defect.
EXCESS Nausea, vomiting, diarrhea, fatigue, calcium
oxalate nephrolithiasis (excess oxalate from
vitamin C metabolism). Can iron toxicity in
predisposed individuals by increasing dietary
iron absorption (ie, can worsen hemochromatosis
or transfusion-related iron overload).
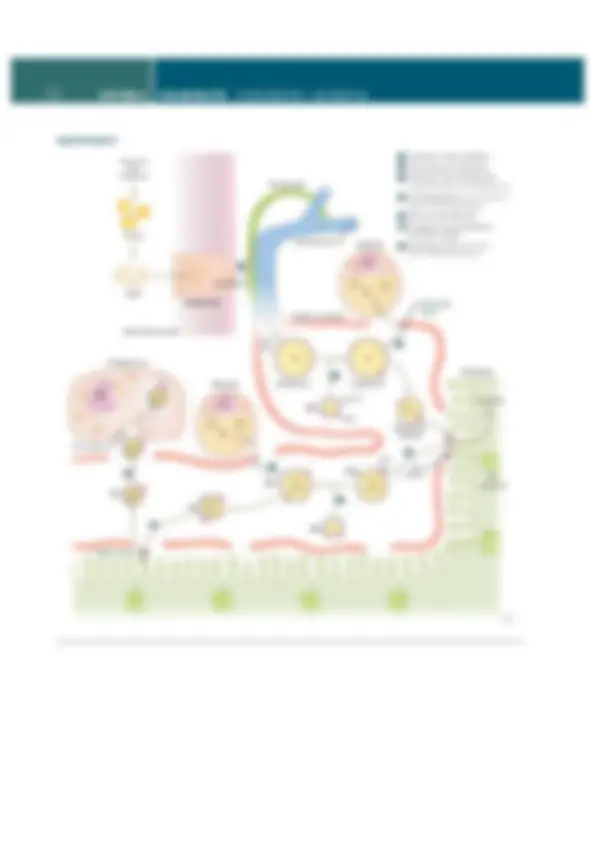
Methionine synthase
THF Methionine
CH
3
to anabolic
pathways
Adenosine
Homocysteine
S-adenosyl
homocysteine
Cysteine
B
12
B
6
THF–CH
3
SAM
Protein Fatty acids with odd number of
carbons, branched-chain amino acids
Methylmalonyl-CoA
mutase
Methylmalonyl-CoA
Succinyl-CoA
TCA cycle
B
12
B
6
Heme
BIOCHEMISTRY ` BIOCHEMISTRY—NUTRITION SEC TION II
Vitamin K Includes phytomenadione, phylloquinone, phytonadione, menaquinone.
FUNCTION Activated by epoxide reductase to the
reduced form, which is a cofactor for the
γ-carboxylation of glutamic acid residues on
various proteins required for blood clotting.
Synthesized by intestinal microbiota.
K is for K oagulation. Necessary for the
maturation of clotting factors II, VII, IX,
X, and proteins C and S. Warfarin inhibits
vitamin K–dependent synthesis of these factors
and proteins.
DEFICIENCY Neonatal hemorrhage with PT and aPTT
but normal bleeding time (neonates have
sterile intestines and are unable to synthesize
vitamin K). Can also occur after prolonged use
of broad-spectrum antibiotics or hepatocellular
disease.
Not in breast milk; “breast-fed infants D on’t
K now about vitamins D and K ”. Neonates are
given vitamin K injection at birth to prevent
hemorrhagic disease of the newborn.
Zinc
FUNCTION Mineral essential for the activity of 100+ enzymes. Important in the formation of zinc fingers
(transcription factor motif).
DEFICIENCY
A
Delayed wound healing, suppressed immunity, male hypogonadism, adult hair (axillary, facial,
pubic), dysgeusia, anosmia. Associated with acrodermatitis enteropathica A (congenital defect in
intestinal zinc absorption manifesting with triad of hair loss, diarrhea, and inflammatory skin rash
around body openings (periorificial) and tips of fingers/toes (acral). May predispose to alcoholic
cirrhosis.
Protein-energy malnutrition
Kwashiorkor Protein malnutrition resulting in skin lesions,
edema due to plasma oncotic pressure (due
to low serum albumin), liver malfunction
(fatty change due to apolipoprotein synthesis
and deposition). Clinical picture is small child
with swollen abdomen A.
Kwashiorkor results from protein-
deficient MEALS :
M alnutrition
E dema
A nemia
L iver (fatty)
S kin lesions (eg, hyperkeratosis,
dyspigmentation)
A B
Marasmus Malnutrition not causing edema. Diet is
deficient in calories but no nutrients are
entirely absent.
M arasmus results in m uscle wasting B.
Linear growth maintained in acute protein-
energy malnutrition (vs chronic malnutrition).
SEC TION II BIOCHEMISTRY ` BIOCHEMISTRY—NUTRITION
Ethanol metabolism
Alcohol dehydrogenase Acetaldehyde dehydrogenase
Catalase
Fomepizole
Disulfiram
Peroxisome
Mitochondria
Cytosol
Microsome
NADH
NADPH
ROS
NADP
Ethanol Acetaldehyde Acetate
H
2
O
NAD
NAD
NADH
CYP2E
H
2
O
2
NADH/NAD
ratio inhibits
TCA cycle
acetyl-CoA used
in ketogenesis ( ketoacidosis),
lipogenesis (
hepatosteatosis).
Females are more susceptible than
males to effects of alcohol due
to activity of gastric alcohol
dehydrogenase, body size,
percentage of water in body
weight.
NAD
is the limiting reagent.
Alcohol dehydrogenase operates via
zero-order kinetics.
Acetyl-CoA
Gluconeogenesis
Glucose
Glycolysis
Lipogenesis
Ketogenesis
Glyceraldehyde-3-P
PEP
Pyruvate
Pathways stimulated by ↑ NADH/NAD
ratio
TCA cycle
↑ Malate
Succinyl-
CoA
OAA
Isocitrate
α-KG
DHAP
↑ Lactate
(anion gap metabolic acidosis)
↑ Fatty acids
(fasting
hypoglycemia)
(hepatic
steatosis)
↑ Ketoacids
↑ Triglycerides
↑ Glycerol-3-P
NADH NAD
NADH
NADH
NAD
NAD
NAD
NADH
OAA
↑
Pathways inhibited by ↑ NADH/NAD
ratio
NADH
NAD
S
Q
R
4 B
4 A
Ethanol metabolism NADH/
NAD
ratio in liver, causing:
Lactic acidosis— pyruvate
conversion to lactate
Fasting hypoglycemia—
gluconeogenesis due to
conversion of OAA to malate
Ketoacidosis—diversion of
acetyl-CoA into ketogenesis
rather than TCA cycle
Hepatosteatosis— conversion
of DHAP to glycerol-3-P
4A ; acetyl-CoA diverges into
fatty acid synthesis 4B , which
combines with glycerol-3-P to
synthesize triglycerides
Fome pizole—competitive inhibitor
of alcohol dehydrogenase;
preferred antidote f or o verdoses
of m ethanol or e thylene glycol.
Alcohol dehydrogenase has
higher affinity for ethanol
than for methanol or ethylene
glycol
ethanol can be used as
competitive inhibitor of alcohol
dehydrogenase to treat methanol
or ethylene glycol poisoning.
Dis ulfiram—blocks acetaldehyde
dehydrogenase
acetaldehyde
hangover symptoms
dis couraging drinking.
SEC TION II BIOCHEMISTRY ` BIOCHEMISTRY—METABOLISM
Metabolism sites
Mitochondria Fatty acid oxidation (β-oxidation), acetyl-CoA production, TCA cycle, oxidative phosphorylation,
ketogenesis.
Cytoplasm Glycolysis, HMP shunt, and synthesis of cholesterol (SER), proteins (ribosomes, RER), fatty acids,
and nucleotides.
Both H eme synthesis, u rea cycle, g luconeogenesis. Hug s take two (both).
Summary of pathways
T
B
T
T
B
B
Glycogen
UDP-glucose Glucose- 1 -phosphate
Glucose
Glucose-6-phosphate 6-phosphogluconolactone
Fructose-6-phosphate
Fructose-1,6-bisphosphate
Glyceraldehyde-3-P DHAP
1,3-bisphosphoglycerate
3-phosphoglycerate
2 -phosphoglycerate
Phosphoenolpyruvate (PEP)
Alanine Pyruvate
Acetyl-CoA
Glyceraldehyde
Ribulose-5-phosphate
Fructose-1-phosphate Fructose
NH
3
2
Carbamoyl phosphate
Citrulline
Aspartate
Argininosuccinate
Urea cycle
Ornithine
Urea
H 2
O
Arginine
Fumarate
Oxaloacetate
Malate
TCA cycle
Succinate
Citrate
Isocitrate
α-ketoglutarate
Succinyl-CoA Methylmalonyl-CoA Propionyl-CoA
Odd-chain fatty acids,
isoleucine, valine, methionine,
threonine, pyrimidines
Acetoacetate
β-hydroxybutyrate
Mevalonate
Galactose
Galactose-1-phosphate
HMP shunt
Fructose metabolism
Lipid metabolism
Galactose metabolism
Gluconeogenesis
Ketogenesis
Glycolysis
Protein metabolism
Glycogenesis / glycogenolysis
Lactate
Acetoacetyl-CoA HMG-CoA
Malonyl-CoA
Triglycerides
Glycerol
Cholesterol
B
12
Irreversible, important point of regulation
Requires thiamine cofactor (TPP)
Requires biotin cofactor
B
T
Fatty acids
Hexokinase/glucokinase
Glucose-6-phosphatase
(von Gierke disease)
Glucose-6-phosphate
dehydrogenase
Transketolase
Pyruvate kinase
Pyruvate dehydrogenase
HMG-CoA reductase
Pyruvate carboxylase
PEP carboxykinase
Citrate synthase
Triose phosphate isomerase
Phosphofructokinase- 1
Fructose-1,6-bisphosphatase 1
Fructokinase (essential fructosuria)
Aldolase B (fructose intolerance)
Aldolase B (liver) , A (muscle)
Isocitrate dehydrogenase
α-ketoglutarate dehydrogenase
Ornithine transcarbamylase
Propionyl-CoA carboxylase
Carbamoyl phosphate synthetase I
Galactokinase (mild galactosemia)
Galactose- 1 -phosphate
uridyltransferase
(severe galactosemia)
BIOCHEMISTRY ` BIOCHEMISTRY—METABOLISM SEC TION II
Activated carriers CARRIER MOLECULE CARRIED IN ACTIVATED FORM
ATP Phosphoryl groups
NADH, NADPH, FADH
2
Electrons
CoA, lipoamide Acyl groups
Biotin CO
2
Tetrahydrofolates 1-carbon units
S-adenosylmethionine (SAM) CH 3
groups
TPP Aldehydes
Universal electron
acceptors
Nicotinamides (NAD
, NADP
from vitamin B 3
and flavin nucleotides (FAD from vitamin B
2
NAD
is generally used in catabolic processes to
carry reducing equivalents away as NADH.
NADPH is used in anabolic processes (eg,
steroid and fatty acid synthesis) as a supply of
reducing equivalents.
NADPH is a product of the HMP shunt.
NADPH is used in:
Anabolic processes
Respiratory burst
Cytochrome P-450 system
Glutathione reductase
Hexokinase vs
glucokinase
Phosphorylation of glucose to yield glucose-6-phosphate is catalyzed by glucokinase in the liver and
hexokinase in other tissues. Hexokinase sequesters glucose in tissues, where it is used even when
glucose concentrations are low. At high glucose concentrations, glucokinase helps to store glucose
in liver. Glucokinase deficiency is a cause of maturity onset diabetes of the young (MODY) and
gestational diabetes.
Hexokinase Glucokinase
Location Most tissues, except liver
and pancreatic β cells
L iver, β cells of pancreas
K
m
Lower ( affinity) Higher ( affinity)
V
max
Lower ( capacity) Higher ( capacity)
Induced by insulin No Yes
Feedback inhibition by Glucose-6-phosphate Fructose-6-phosphate
BIOCHEMISTRY ` BIOCHEMISTRY—METABOLISM SEC TION II
Pyruvate
dehydrogenase
complex deficiency
Causes a buildup of pyruvate that gets shunted to lactate (via LDH) and alanine (via ALT).
X-linked.
FINDINGS Neurologic defects, lactic acidosis, serum alanine starting in infancy.
TREATMENT intake of ketogenic nutrients (eg, high fat content or lysine and leucine).
Pyruvate metabolism Functions of different pyruvate metabolic
pathways (and their associated cofactors):
Alanine aminotransferase (B
6
): alanine
carries amino groups to the liver from
muscle
Pyruvate carboxylase (B
7
): oxaloacetate
can replenish TCA cycle or be used in
gluconeogenesis
Pyruvate dehydrogenase (B 1
, B
2
, B
3
, B
5
lipoic acid): transition from glycolysis to
the TCA cycle
Lactic acid dehydrogenase (B 3
): end of
anaerobic glycolysis (major pathway in
RBCs, WBCs, kidney medulla, lens,
testes, and cornea)
Lactate
Mitochondria
Cytosol
NADH
NAD
NADH
Acetyl-CoA
NAD
CO
2
CO
2
AL
T
LDH
PC
PDH
Alanine
Cahill cycle
Cori cycle
Oxaloacetate
Glucose
Pyruvate
TCA cycle
Acetyl-CoA (2C)
NADH
Malate (4C)
Fumarate (4C)
Succinate (4C)
Succinyl-
CoA (4C)
α-KG (5C)
Isocitrate (6C)
cis -Aconitate
Citrate (6C)
GTP + CoA
Oxalo-
acetate
(4C)
Pyruvate (3C)
PDH
Succinyl-CoA
NADH
ATP
ATP
NADH
ADP
ATP
Acetyl-CoA
NADH
ATP
CO
2
CO
2
CO
2
FADH
2
α
K
G
d
e
h
y
d
r
o
g
e
n
a
s
e
C
i t
r a t e
s y n t h a s e
Isocitrate
dehydrogenase
Also called Krebs cycle. Pyruvate acetyl-CoA
produces 1 NADH, 1 CO 2
The TCA cycle produces 3 NADH, 1 FADH
2
2 CO
2
, 1 GTP per acetyl-CoA = 10 ATP/
acetyl-CoA (2× everything per glucose). TCA
cycle reactions occur in the mitochondria.
α-ketoglutarate dehydrogenase complex
requires the same cofactors as the pyruvate
dehydrogenase complex (vitamins B 1
, B
2
, B
3
B
5
, lipoic acid).
C itrate i s K rebs’ s tarting s ubstrate f or m aking
o xaloacetate.
SEC TION II BIOCHEMISTRY ` BIOCHEMISTRY—METABOLISM
Electron transport
chain and oxidative
phosphorylation
NADH electrons are transferred to complex I.
FADH
2
electrons are transferred to complex II
(at a lower energy level than NADH).
The passage of electrons results in the formation
of a proton gradient that, coupled to oxidative
phosphorylation, drives ATP production. ATP
hydrolysis can be coupled to energetically
unfavorable reactions.
Uncoupling proteins (found in brown fat, which
has more mitochondria than white fat) produce
heat by inner mitochondrial membrane
permeability
proton gradient. ATP synthesis
stops, but electron transport continues.
1 NADH 2.5 ATP; 1 FADH
2
1.5 ATP
NADH electrons from glycolysis enter
mitochondria via the malate-aspartate or
glycerol-3-phosphate shuttle.
Aerobic metabolism of one glucose molecule
produces 32 net ATP via malate-aspartate
shuttle (heart and liver), 30 net ATP via
glycerol-3-phosphate shuttle (muscle).
Anaerobic glycolysis produces only 2 net ATP
per glucose molecule.
Aspirin overdose can also cause uncoupling
of oxidative phosphorylation resulting in
hyperthermia.
Mitochondrial
matrix
Inner mitochondrial
membrane
Intermembrane
space
Complex I Complex II
(succinate
dehydrogenase)
Complex III Complex IV
Cyanide,
CO
Complex V
ADP + P
i
NADH
Uncoupling proteins
Aspirin overdose
NAD
H
H
H
H
CoQ
Cyto-
chrome c
FADH
2
FAD 1/2 O 2
H
2
O
ATP
Gluconeogenesis,
irreversible enzymes
All enzymes may be subject to activation by
glucagon in fasting state.
P athway p roduces f resh g lucose.
Pyruvate carboxylase In mitochondria. Pyruvate oxaloacetate.
Requires biotin, ATP. Activated by acetyl-CoA.
Phosphoenolpyruvate
carboxykinase
In cytosol. Oxaloacetate
phosphoenolpyruvate (PEP).
Requires GTP.
Fructose-1,6-
bisphosphatase 1
In cytosol. Fructose-1,6-bisphosphate
fructose-6-phosphate.
Citrate
⊕ , AMP
⊝ , fructose 2,6-bisphosphate
⊝ .
Glucose-6-
phosphatase
In ER. Glucose-6-phosphate
glucose.
Occurs primarily in liver; serves to maintain euglycemia during fasting. Enzymes also found in
kidney, intestinal epithelium. Deficiency of the key gluconeogenic enzymes causes hypoglycemia.
(Muscle cannot participate in gluconeogenesis because it lacks glucose-6-phosphatase).
Odd -chain fatty acids yield 1 propionyl-CoA during metabolism, which can enter the TCA cycle
(as succinyl-CoA), undergo gluconeogenesis, and serve as a glucose source (It’s odd for fatty acids
to make glucose ). Even-chain fatty acids cannot produce new glucose, since they yield only acetyl-
CoA equivalents.
SEC TION II BIOCHEMISTRY ` BIOCHEMISTRY—METABOLISM
Disorders of fructose metabolism
Essential fructosuria Hereditary fructose intolerance
ENZYME DEFICIENCY Fructokinase (autosomal recessive) Aldolase B (autosomal recessive)
PATHOPHYSIOLOGY Fructose is not trapped into cells. Hexokinase
becomes 1° pathway for converting fructose to
fructose-6-phosphate.
Fructose-1-phosphate accumulates
available
phosphate inhibition of glycogenolysis and
gluconeogenesis.
PRESENTATION (SIGNS/SYMPTOMS) Asymptomatic, benign. Fructose appears in
blood and urine (fructo kin ase deficiency is
kin der).
Hypoglycemia, jaundice, cirrhosis, vomiting.
Symptoms only present following consumption
of fruit, juice, or honey.
ADDITIONAL REMARKS Urine dipstick will be
⊝ (tests for glucose only); reducing sugar can be detected in the urine
(nonspecific test for inborn errors of carbohydrate metabolism).
TREATMENT – intake of fructose, sucrose (glucose + fructose),
and sorbitol (metabolized to fructose).
Fructokinase Aldolase B
Dihydroxyacetone-P
Glyceraldehyde
Glyceraldehyde-3-P
Glycerol
NADH
Triose kinase
ATP
ADP
ATP
ADP
NAD
Fructose Fructose-1-P
Glycolysis
Triose phosphate
isomerase
Disorders of galactose metabolism
Galactokinase deficiency Classic galactosemia
ENZYME DEFICIENCY Galactokinase (autosomal recessive). Galactose-1-phosphate uridyltransferase
(autosomal recessive).
PATHOPHYSIOLOGY Galactitol accumulates if diet has galactose. Damage caused by accumulation of toxic
substances (eg, galacitol).
PRESENTATION (SIGNS/SYMPTOMS) Relatively mild/benign condition (galacto kin ase
deficiency is kin der).
Galactose appears in blood (galactosemia) and
urine (galactosuria); infantile cataracts. May
present as failure to track objects or develop
social smile.
Symptoms start when infant is fed formula
or breast milk
failure to thrive, jaundice,
hepatomegaly, infantile cataracts (galacitol
deposition in eye lens), intellectual disability.
Can predispose neonates to E coli sepsis.
TREATMENT – Exclude galactose and lactose (galactose +
glucose) from diet.
Galactose Galactose-1-P
Galactokinase
Aldose
reductase
Galactitol
Uridylyltransferase
4-Epimerase Glycolysis/glycogenesis
Glucose-1-P
ATP
ADP
UDP-Glu UDP-Gal
BIOCHEMISTRY ` BIOCHEMISTRY—METABOLISM SEC TION II
Sorbitol An alternative method of trapping glucose in the cell is to convert it to its alcohol counterpart,
sorbitol, via aldose reductase. Some tissues then convert sorbitol to fructose using sorbitol
dehydrogenase; tissues with an insufficient amount/activity of this enzyme are at risk of
intracellular sorbitol accumulation, causing osmotic damage (eg, cataracts, retinopathy, and
peripheral neuropathy seen with chronic hyperglycemia in diabetes).
High blood levels of galactose also result in conversion to the osmotically active galactitol via aldose
reductase.
L iver, o varies, and se minal vesicles have both enzymes (they lose sorbitol).
Glucose
NADPH NAD
Aldose reductase
Sorbitol
Sorbitol dehydrogenase
Fructose
L ens has primarily A ldose reductase. R etina, K idneys, and S chwann cells have only aldose
reductase ( LARKS ).
Lactase deficiency Insufficient lactase enzyme dietary lactose intolerance. Lactase functions on the intestinal brush
border to digest lactose (in milk and milk products) into glucose and galactose.
Primary: age-dependent decline after childhood (absence of lactase-persistent allele), common in
people of Asian, African, or Native American descent.
Secondary: loss of intestinal brush border due to gastroenteritis (eg, rotavirus), autoimmune disease.
Congenital lactase deficiency: rare, due to defective gene.
Stool demonstrates pH and breath shows hydrogen content with lactose hydrogen breath test
(H
is produced when colonic bacteria ferment undigested lactose). Intestinal biopsy reveals
normal mucosa in patients with hereditary lactose intolerance.
FINDINGS Bloating, cramps, flatulence (all due to fermentation of lactose by colonic bacteria
gas), and
osmotic diarrhea (undigested lactose).
TREATMENT Avoid dairy products or add lactase pills to diet; lactose-free milk.
Amino acids Only l-amino acids are found in proteins.
Essential PVT TIM H a LL : P henylalanine, V aline, T ryptophan, T hreonine, I soleucine, M ethionine,
H istidine, L eucine, L ysine.
Glucogenic: Met hionine, his tidine, val ine. We met his val entine, who is so sweet ( gluco genic).
Glucogenic/ketogenic: Isoleucine, phenylalanine, threonine, tryptophan.
Ketogenic: l eucine, l ysine. The on l y pure l y ketogenic amino acids.
Acidic Aspartic acid , glutamic acid.
Negatively charged at body pH.
Basic Ar ginine, his tidine, lys ine.
Arginine is most basic. Histidine has no charge at body pH.
Arginine and histidine are required during periods of growth.
Arginine and lysine are in histones which bind negatively charged DNA.
His lys (lies) ar e basic.
BIOCHEMISTRY ` BIOCHEMISTRY—METABOLISM SEC TION II
Ornithine
transcarbamylase
deficiency
Most common urea cycle disorder. X-linked recessive (vs other urea cycle enzyme deficiencies,
which are autosomal recessive). Interferes with the body’s ability to eliminate ammonia. Often
evident in the first few days of life, but may present later. Excess carbamoyl phosphate is converted
to orotic acid (part of the pyrimidine synthesis pathway).
Findings: orotic acid in blood and urine, BUN, symptoms of hyperammonemia. No
megaloblastic anemia (vs orotic aciduria).
Amino acid derivatives
Tryptophan
Niacin NAD
/NADP
Serotonin Melatonin
Phenylalanine NE
Thyroxine
Tyrosine Dopa Dopamine
Histidine Histamine
Glycine Porphyrin Heme
Epi
Arginine
BH
4
= tetrahydrobiopterin
Urea
Nitric oxide
Creatine
Melanin
B
2
, B
6
BH
4
, B
6
BH
4
BH
4
BH
4
SAM B
6
Vitamin C
B
6
B
6
Glutathione
Glutamate
B GABA
6
B 6
Catecholamine synthesis/tyrosine catabolism
Phenylalanine
BH 4
BH 4
Tyrosine
B
6
Vitamin C
SAM
Dopamine
Norepinephrine
Epinephrine Metanephrine
Normetanephrine
Vanillylmandelic acid
Monoamine
oxidase
Monoamine
oxidase
Homovanillic acid
Homogentisic acid
Maleylacetoacetic acid
Phenylalanine
hydroxylase
PKU
Tyrosinase
Tyrosine
hydroxylase
Melanin
Cortisol
Albinism
TCA cycle
Alkaptonuria
Homogentisate
oxidase
DOPA
decarboxylase
Dopamine
β-hydroxylase
Carbidopa
Catechol-O-methyltransferase
Catechol-O-
methyltransferase
DOPA
(Dihydroxyphenylalanine)
Fumarate
Phenylethanolamine- N-
methyltransferase
SEC TION II BIOCHEMISTRY ` BIOCHEMISTRY—METABOLISM
Phenylketonuria Caused by phenylalanine hydroxylase (PAH).
Tyrosine becomes essential. phenylalanine
phenyl ketones in urine.
Tetrahydrobiopterin (BH 4
) deficiency—BH 4
essential cofactor for PAH. BH
4
deficiency
phenylalanine. Varying degrees of clinical
severity. Untreated patients typically die in
infancy.
Phenylalanine embryopathy— phenylalanine
levels in pregnant patients with untreated
PKU can cause fetal growth restriction,
microcephaly, intellectual disability, congenital
heart defects. Can be prevented with dietary
measures.
Autosomal recessive.
Screening occurs 2–3 days after birth (normal at
birth because of maternal enzyme during fetal
life).
Findings: intellectual disability, microcephaly,
seizures, hypopigmented skin, eczema, musty
body odor.
Treatment: phenylalanine and tyrosine in
diet (eg, soy products, chicken, fish, milk),
tetrahydrobiopterin supplementation.
Phenyl ketones—phenylacetate, phenyllactate,
and phenylpyruvate.
Disorder of aromatic amino acid metabolism
musty body odor.
Patients with PKU must avoid the artificial
sweetener aspartame, which contains
phenylalanine.
Dietary protein
Aspartame
Endogenous
protein
Phenyl ketones
Phenylalanine
BH₄ BH₂
Dihydropteridine
reductase
Phenylalanine
hydroxylase
PKU
Tetrahydrobiopterin
deficiency
Tyrosine
Thyroxine
Dopamine
Melanin
Norepinephrine/epinephrine
NAD
NADH + H
Maple syrup urine
disease
Blocked degradation of branched amino acids
( I soleucine, l eucine, v aline) due to branched-
chain α-ketoacid dehydrogenase (B
1
). Causes
α-ketoacids in the blood, especially those of
leucine.
Treatment: restriction of isoleucine, leucine,
valine in diet, and thiamine supplementation.
Autosomal recessive.
Presentation: vomiting, poor feeding, urine
smells like maple syrup/burnt sugar. Causes
progressive neurological decline.
I l ove V ermont maple syrup from maple trees
(with B
1
ranches ).
Alkaptonuria
A
Congenital deficiency of homogentisate oxidase in the degradative pathway of tyrosine to fumarate
pigment-forming homogentisic acid builds up in tissue. Autosomal recessive. Usually benign.
Findings: bluish-black connective tissue, ear cartilage, and sclerae (ochronosis A
); urine turns
black on prolonged exposure to air. May have debilitating arthralgias (homogentisic acid toxic to
cartilage).