Download Bonding in Molecules and more Slides Geometry in PDF only on Docsity!
Bonding in Molecules
Michaelmas Term - Second Year 2019
These 8 lectures build on material presented in “Introduction to Molecular Orbitals” (HT Year 1). They provide a basis for
analysing the shapes, properties, spectra and reactivity of a wide range of molecules and transition metal compounds.
The essentials of molecular orbital theory
- The requirements for a good theory of bonding
- The orbital approximation
- The nature of molecular orbitals
- The linear combination of atomic orbitals (LCAO) approach to molecular orbitals
Diatomic molecules: H 2
+
, H
2
and AH
- The wave functions for H 2
+
and H 2
using an LCAO approach
- MO schemes for AH molecules (A = second period atom, Li to F)
Symmetry and molecular orbital diagrams for the first row hydrides AH n
- The use of symmetry in polyatomic molecules
- MO treatment of AH 2
( C 2v
)
- MO diagrams for AH 3
( C 3v
)
- MO diagrams for AH 4
( T d
)
Photoelectron spectroscopy and experimental energy levels
- Photoelectron spectroscopy and "experimental" MO diagrams
- Photoelectron spectra of AH n
molecules
The use of Walsh diagrams in exploring molecular shapes
- The shapes of AH 2
molecules
- The bonding and shapes of H 3
+
and H 3
-
: 3c-2e and 3c-4e bonds
Molecular orbital diagrams for hyper-coordinate molecules
- The bonding in XeF 2
(and CO 2
)
- 12-electron main group octahedral systems: SF 6
as an example
- Expanding the coordination sphere in carbon: [C(AuPR 3
) 6
]
2+
as an analogue of CH 6
2+
Fragment approach to bonding in electron deficient clusters
- Build up of molecules from fragments
- Bonding in [B 6
H 6
]
2–
(from 6 equivalent BH fragments) and Wade’s rules , the concept of isolobality
Complexes of the transition metals: octahedral, tetrahedral and square planar.
- Octahedral transition metal complexes: σ−bonding
- π-interactions and the spectrochemical series
- Molecular orbitals for 4-coordinate geometries: ML 4
( T d
and D 4h
)
- A miscellany of bonds (quadruple, quintuple, sextuple!)
Bibliography
- Jean, Volatron and Burdett; An Introduction to Molecular Orbitals (OUP)
- DeKock and Gray: Chemical Structure and Bonding (Benjamin)
- Burdett: Molecular Shapes (Wiley)
- Burdett: The Chemical Bond: A Dialog (Wiley).
- Albright, Burdett and Whangbo: Orbital Interactions in Chemistry (Wiley)
- Streitweiser: Molecular orbital theory for Organic Chemists (Wiley)
- Albright and Burdett: Problems in Molecular Orbital Theory (OUP)
Resources
Character tables
http://global.oup.com/uk/orc/chemistry/qchem2e/student/tables/
Software:
Python_extended_huckel: http://course.chem.ox.ac.uk/bonding-in-molecules-year-2-2019.aspx
and a python (3.x) platform to run it:
Anaconda https://www.continuum.io/downloads
atomic orbital or another influences the resultant molecular orbitals and (consequently) the molecular
properties.
In the simplest form of LCAO theory, only the valence orbitals of the atoms are used to construct
MOs (e.g. just the 1 � for hydrogen, only the 2 � and 2 � for carbon, and so on). In more accurate forms
of calculation other orbitals are also included (see below in the treatment of H 2
). The atomic orbitals
used are known as a basis set. In general, the larger the basis set the more accurate are the
calculations (e.g. of bond energies and distances). The down-side is that the calculations become very
computationally demanding as the size of the basis set increases, and in general only the "simplest"
systems can be calculated with a great degree of accuracy.
Linear combinations of atomic orbitals (LCAOs) are used to construct molecular orbitals � �
. If we
take two AOs, � �
and �
�
, of energies −�
�
and −�
�
respectively, we form two molecular orbitals:
�
�
�
�
�
�
�
�
�
�
The MOs form an orthonormal set. This comprises two key features, namely:
the normalization condition: ∫
�
�
and the orthogonality condition: ∫
�
�
Overlap
Atomic orbitals must overlap in order to combine to form an MO. In other words, the overlap
integral � � �
between two atomic orbitals �
�
and �
�
must be non-zero.
� �
�
�
In diatomics, it is relatively straightforward to see which orbitals overlap and which do not:
In polyatomic molecules (see Section 7 below), it is not so easy to see, and we need to use group
theory. The key result from group theory is that only orbitals carrying the same symmetry labels
(when analysed in the same point group) can overlap, otherwise � � �
is zero.
Orbitals which overlap constructively ( i.e. "in-phase") are bonding. Orbitals which overlap
destructively are antibonding. In general, the greater number of internuclear nodes the higher the
energy of an orbital ( c.f. AOs in the H atom, particle in a box, etc .).
(but see Baird, J Chem Ed 1986 , 63 , 663 for a discussion of the origin of bonding).
The energy of stabilization of a bonding MO, and the energy of destabilization of an anti-bonding MO
(with respect to the combining AOs) depends on:
� �
, between the AOs and
- The energy separation of the combining AOs , Δ� = |�
�
�
|. The energy of stabilisation
of an MO is at a maximum when the two combining AOs have similar energy. Conversely, orbitals
with very different energies will interact poorly, irrespective of the size of the overlap.
To ascertain the best possible wave function within the LCAO approximation, we use the variation
principle. The variation principle states that for any trial wavefunction � � � � � �
� � � �
� � � �
is the
true energy of the system).
For a trial wavefunction, � � � � � �
, the associated energy is given by:
���� �
� � � � �
∗
� � � � �
� � � � �
∗
� � � � �
The task is to find the set of coefficients � �
in the molecular orbitals �
�
�
�
�
�
give the minimum possible energy ( i.e. we vary them until the change is less than a target criterion).
There are a number of well-established computational procedures for doing this.
6. Heteronuclear diatomics AH molecules (A = second period atom, Li to F)
We shall first consider linear heteronuclear diatomic molecules AH where A is one of the second
period atoms, Li to F. These atoms have one 2� and three 2� valence orbitals. In such systems it is
conventional to take z as the molecular axis (as is also the case for homonuclear diatomics A 2
Symmetry analysis: Pt grp C ∞ v
From the group theory (character) table for C ∞ v
(see appendix):
For the A atom (4 AOs): Γ (2� ) = σ Γ (2�
�
) = σ Γ (2�
� ,�
) = π
For the H atom (1 AO): Γ (1s) = σ
For the AH molecule (5 MOs): Γ (MOs) = 3σ + 1π (π is a doubly degenerate set)
The valence 2� and 2� �
atomic orbitals of the A atom overlap with the H 1 s orbital (all are σ
symmetry). The 2�
� ,�
(π) have no symmetry match on H, so will be non-bonding. The three MOs of σ
symmetry take the form
�
� � (� )
�
� � (� )
�
� �
�
(� )
In all cases, one σ bonding, one σ-non-bonding and one σ anti-bonding orbital are formed, but the
distribution of bonding/non-bonding/antibonding character between the three MOs varies.
Perturbation theory tells us that the interaction between two orbitals with different energies depends
on the overlap (squared) divided by the energy difference between them (
2
S
E
Figure: Interaction energies between two orbitals with different ∆E.
Even if overlap is good, when orbitals have very different energies their interaction will be very
weak and they will be effectively non-bonding.
Note that the MOs are labelled according to their symmetry. For orbitals of the same symmetry, the
lowest in energy is labelled 1 (here 1σ), the next 2 (2σ), etc.
In HB the bonding is maximized in the more stable orbital (i.e. the 1σ level above). The mixing of the
s and p orbitals in the MO of a molecule is the equivalent of hybridization in valence bond theory. 2�
- 2� mixing results in a build up (as in 1σ) or diminution (as in 2σ) of orbital overlap and electron
density in the internuclear region (similar to N 2
, see Year 1 notes).
In the case of HF the 2� orbital on F is too low in energy to interact significantly with the 1 s orbital,
and the 1a 1
MO is almost entirely made up from the 2� atomic orbital of the A atom. In other words,
�
�
�
| in the molecular orbital
�
�
�
�
and the lower energy orbital (1σ) now behaves like a core orbital.
If the A atom is very electropositive (Li, Be) the 2� and
2� valence orbitals lie high in energy and above that of
the H 1 s orbital (note 2� /2� separation is due to
penetration). If, on the other hand, A is very
electronegative ( e.g. F) its 2� and 2� orbitals lie lower
in energy than H 1 s. For elements of intermediate
electronegativity, such as B, C and N, the H 1 s orbital
lies between the A atom's 2� and 2� levels (the valence
shell energy of the H atom 1 s orbital is normally taken
as -13.6 eV). In general we expect much more s - p
mixing in B and C than in O and F because the energy
difference between 2� and 2� levels is not yet
prohibitively large.
Note that it is widespread practice (and generally convenient) to refer to "σ" and "π" type orbitals
even though in a strict (group theoretical) sense these Mulliken symbols for irreducible
representations only formally apply to the cylindrical point groups D ∞ h
and C
∞ v
(even in the latter
instance it is also permitted to use the a 1
, e
1
etc. symbols). For example, in ethene we like to refer to
H
2
C=CH
2
"σ" and " π" bonds even though the molecular orbitals being referred to actually have the
Mulliken symbols a g
and b
1u
in the D
2h
symmetry of ethene.
Valence shell energies (in eV) of 2 � and 2 � electrons of second period atoms
Li Be B C N O F Ne
Symmetry and molecular orbital diagrams for the first row hydrides AH
n
We have already seen for the AH molecule how knowledge of the symmetry (irreducible
representations) of atomic orbitals helps identify those atomic orbitals that (in principle at least) can
overlap to form MOs. Symmetry becomes an indispensible tool when analysing the bonding in
polyatomic molecules AH n
with n > 1.
7. The use of symmetry in polyatomic molecules
So far we have a good idea of the MO pattern when 2 or 3 AOs interact. With polyatomic systems we
have a potentially very large number of AOs, so expect an equally large number of MOs. However if a
molecule is symmetric (or can be approximated as such) we can still deal with their interactions in a semi-
qualitative way.
If two or more atoms are chemically identical, they must be associated with equal electron density.
In H 2
we have:
�
�
�
�
but electron density (given by �
�
) must be the same on the two indistinguishable atoms.
Hence:
�
�
�
�
�
�
Therefore in any molecule with two equivalent H atoms (H 2
O, HC≡CH etc ) the contribution to any MO in
the molecule will be either � �
�
�
or �
�
�
�
Such linear combinations of equivalent
atomic orbitals are known as Symmetry Adapted Linear Combinations (SALCs).
If we can generate SALCs for equivalent orbitals for molecules just by using symmetry, we pre-determine
the ratio of coefficients of the AOs in the MOs and the interaction between the different atoms is then
often reduced to that between 2 or 3 orbital sets. Many examples of this will be presented throughout
these lectures and later courses.
SALCs for various symmetry arrays of equivalent orbitals are given in the appendix, along with relevant
character tables. You should now be familiar with these from the "Symmetry 1" 2nd year lectures.
8. MO treatment of AH 2
( C
2 v
We shall take as our specific example H 2
O, but the general scheme developed will be applicable to
any C 2 v
symmetric AH
2
main group molecule. We will use group theory to classify the irreducible
representation of SALCs of the H atom 1s AOs in the C 2v
symmetry of bent AH
2
. Having then
identified the irreducible representations of the O atom 2 � and 2 � orbitals (in C 2 v
symmetry) we shall
then construct an MO diagram for H 2
O using the principles of AO overlap and energy separation
developed above.
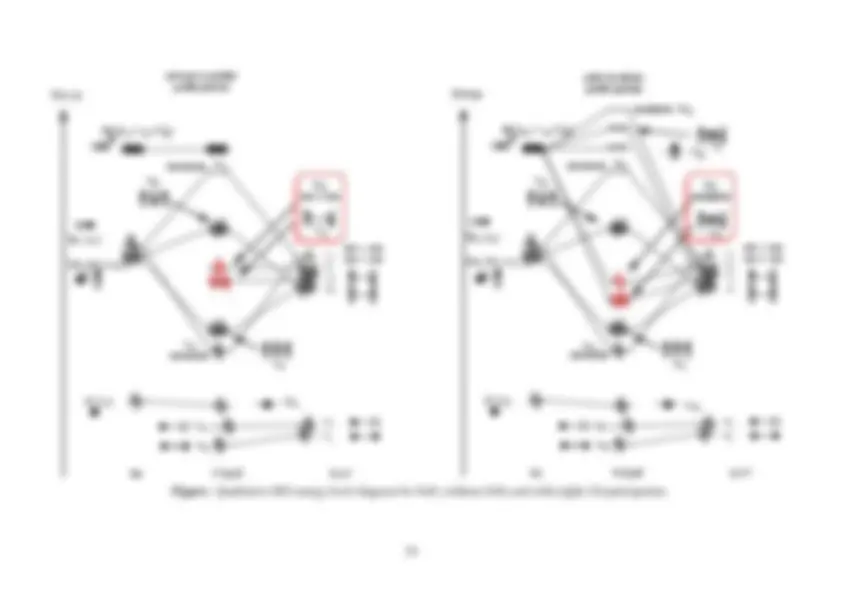
Figure: MO diagrams for H 2
O (cartoon and “real” versions. Note different orders of orbitals 5 and 6.
In this MO diagram the highest occupied molecular orbital (HOMO) is the non-bonding 1b 1
level and
is indicated by the two arrows representing paired electrons. The lowest unoccupied molecular orbital
(LUMO) is the 3a 1
level. Note that (as is the usual convention) we have used C
2v
Mulliken symbols
for the oxygen AOs and (H·····H) SALCs even though the real symmetry of these fragments is much
higher than that of the resultant H 2
O molecule.
A variational calculation for water gives the AO coefficients shown in the Table below. The results show
that there is very little oxygen 2 � �
or H 1s AO contribution in the lowest of the 1a
1
symmetry valence
MOs
Table : MO coefficients for extended Hűckel calculation on H
2
O
Oxygen Hydrogen
MO 2 � 2 �
�
�
�
1 s
1
1 s
2
1a
1 0.91 -0.02 0 0 0.12 0.
2a
1 -0.16 0.93 0 0 0.15 0.
1b
2
1b
1 0 0 0 1 0 0
2b
2 0 0 0.85 0 -0.75 0.
3a
1 0.69 0 0.46 0 -0.77 -0.
The schematic cartoons given in the MO diagram above emphasise the AO contributions and are helpful to
visualise the LCAO origins of the MOs. However, a more accurate picture of an MO function is given by
a contour diagram such as those shown in the ‘real’ figure.
9. MO treatment of AH 3
( C
3v
We shall take as our specific example NH 3
, but again the bonding scheme developed will be broadly
applicable to any C 3v
symmetric AH
3
main group molecule (CH 3
, PH
3
). As above we will use group
theory to classify the irreducible representation of SALCs of the three H atom AOs, along with the
2 � and 2 � AOs of the central N atom in the C 3v
symmetry of the molecule.
Symmetry analysis The coordinate system conventionally chosen has the C 3
axis lying along the
molecular z axis.
The 2 � and 2 � AOs of N and the SALCs of C 3v
(H)
3
span the irreducible representations:
For the N (A) atom (4 AOs): Γ ( 2 � ) = a 1
�
) = a
1
� ,�
) = e
For the H atoms (3 x 1 s AO): Γ (3 x 1 s ) = a 1
Hence for the resulting NH 3
molecule (7 MOs): Γ (MOs) = 3a
1
Note that the combinations derived from 2 �
� ,�
form a degenerate pair because the C
3
operation in C
3v
mixes the 2 �
�
and 2 �
�
AOs so they must have identical energy.
Note that we anticipate no non-bonding MOs in this AH n
compound since all of the carbon atomic
orbitals (a 1
2
) have a symmetry match among the (H)
n
SALCs. In the AH
n
(n = 1, 2, 3) compounds
discussed above symmetry analyses predicted three, two or one non-bonding MOs (AH: 1 x a 1
and 1 x
e 1
non-bonding MOs; AH
2
: 1 x a
1
and 1 x b
1
; AH
3
: 1 x a
1
non-bonding MO).
Note that the combinations derived from the 2�
� ,� ,�
form a triply degenerate set of MOs because the
C
3
operation in the T
d
point group mixes the three 2� AOs, so they must have identical energy.
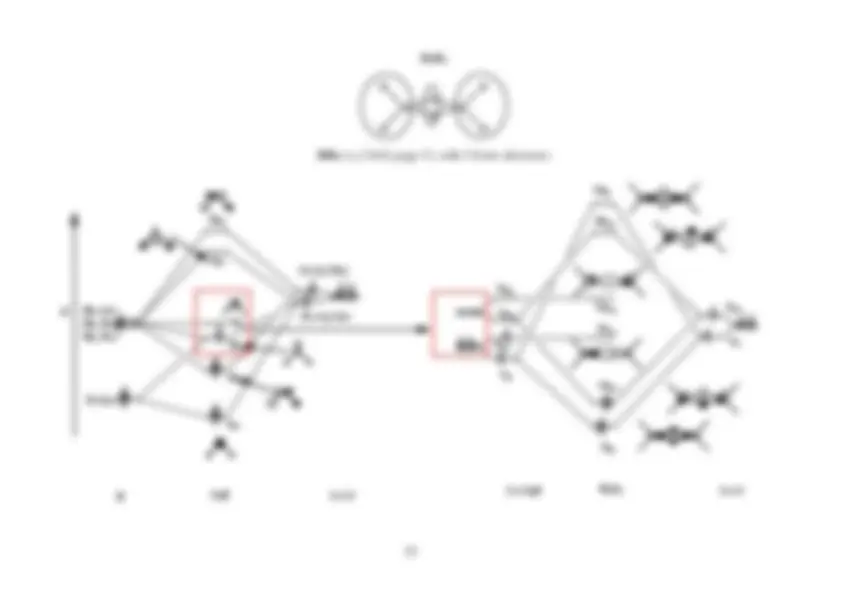
The resultant MO scheme for CH 4
is illustrated below. Note that although there is a favourable 2� -
2� AO energy separation, there is no s - p mixing in CH 4
because the 2� and 2�
� ,� ,�
orbitals have
different irreducible representations in the T d
point group (the overlap, S , is zero, and therefore so is
2
S
∆ E
Figure: LCAO MO scheme for CH
4
11. Photoelectron spectroscopy and experimental energy levels
It is helpful to have some experimental tests of the electronic structures proposed and this is where
photoelectron spectroscopy (PES) has important applications. Just as atomic spectroscopy can give
information on atomic orbitals and their energies, we can obtain information on molecular orbitals by
studying ionization of molecules.
In a photoelectron (PE) experiment, monochromatic radiation (single energy photons) hν, are used to
ionize gas phase molecules, and the kinetic energy (KE) of the ejected electrons is measured.
Einstein’s equation is used to convert the KEs to ionization energies (IEs).
IE = hν - KE
A PE spectrum consists of the number of electrons N (E) of a particular energy plotted against the IEs.
The simplest molecular PE spectrum is that of H 2
. Photoejection of an electron leads to the formation
of H
2
. The PE spectrum of H
2
is very well understood and is reproduced below.
Figure: The photoelectron spectrum of H
2
KE = h ν − IE
PES of HF
Figure: The photoelectron spectrum of HF
As in the spectrum of H 2
, the fine structure in the bands corresponds to the different vibrational states
in which the molecular HF
ion can be formed. Note that the lowest energy state (
2
Π) of the ion is
most likely to be formed in the lowest vibrational energy level. This suggests that there is little
difference between the bond lengths of the ion in this state and the molecule; the electron that has
been removed is therefore non-bonding.
The energy levels deduced from the PE spectrum of HF can readily be interpreted in terms of the
ground state MO description given on Page 9. Clearly we do not expect to get any direct information
about the unoccupied MOs from the PE spectra.
Figure: Interpretation of the PE spectrum of HF in terms of the LCAO MO scheme
12. Photoelectron spectra of AH
n
molecules
Schematic representations of the PE spectra of the isoelectronic series of species: CH 4
, NH
3
, H
2
O, HF
and Ne are shown in the following Figure. The photon energy used is not sufficient to ionize the 1 � core
electrons so for each system we are looking at the ionization of the eight valence electrons.
m
o
s
t
l
y
2
s
m
o
s
t
l
y
2
p
Figure: PE spectra of the isoelectronic series of species: CH
4
, NH
3
, H
2
O, HF and Ne. Energies of the
atomic orbitals are plotted below ( c.f page 10).
The number of bands are related to the symmetry of the species under consideration:
Species Symmetry No. of bands Symmetry labels for orbitals
CH
4
T
d
2 t
2
, a
1
NH
3
C
3v
3 2a
1
, e, 1a
1
H
2
O C
2v
4 b
1
, 2a
1
, b
2
, 1a
1
HF C
∞v
3 π, 2σ, 1σ
Ne R 2 p, s
The energy of the highest IE band is dominated by the energy of the 2 � orbital of the A atom. The
binding energy increases across the series in a similar manner to that of the 2 � orbital. The other orbitals
have an energy variation similar to that of the 2 � orbitals.