Download Spliced Alignment Problem: Solution and Secondary Structure Prediction in RNA - Prof. Saur and more Study notes Computer Science in PDF only on Docsity!
Dynamic Programming
(cont’d)
CS 466
Saurabh Sinha
Spliced Alignment
- Begins by selecting either all putative exons between potential acceptor and donor sites or by finding all substrings similar to the target protein (as in the Exon Chaining Problem).
- This set is further filtered in a such a way that attempt to retain all true exons, with some false ones.
- Then find the chain of exons such that the sequence similarity to the target protein sequence is maximized
The DAG
- Vertices: One vertex for each block in B
- Directed edge connecting non-overlapping blocks
- Label of vertex = string of block it represents
- A path through the DAG spells out the string obtained by concatenating that particular chain of blocks
- Weight of a path is the score of the optimal alignment between the string it spells out and the target sequence
Dynamic programming
- Genomic sequence G = g 1 g 2 …g n
- Target sequence T = t 1 t 2 …t m
- As usual, we want to find the optimal alignment score of the i-prefix of G and the j-prefix of T
- Problem is, there are many i-prefixes possible (since multiple blocks may include position i)
If i is not the starting vertex of block B :
- S(i, j, B) = max { S(i – 1, j, B) – indel penalty S(i, j – 1, B) – indel penalty S(i – 1, j – 1, B) + δ(g i , t j ) } If i is the starting vertex of block B :
- S(i, j, B) = max { S(i, j – 1, B) – indel penalty max all blocks B’ preceding block B S(end(B’), j, B’) – indel penalty max all blocks B ’ preceding block B S(end(B’), j – 1, B’) + δ(g i , t j ) }
RNA secondary structure
prediction
RNA
- There’s more to RNA than mRNA
- RNA can adopt interesting non-linear structures, and catalyze reactions
- tRNAs (transfer RNAs) are the “adapters” that implement translation
Secondary structure
- Several interesting RNAs have a conserved secondary structure (resulting from base- pairing interactions)
- Sometimes, the sequence itself may not be conserved for the function to be retained
- It is important to tell what the secondary structure is going to be, for homology detection
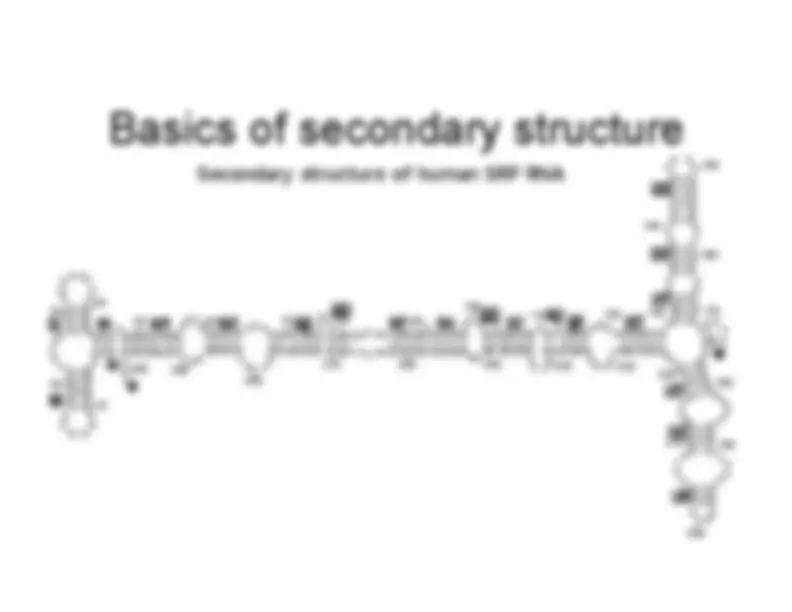
Basics of secondary structure
- G-C pairing: three bonds (strong)
- A-U pairing: two bonds (weaker)
- Base pairs are approximately coplanar
Basics of secondary structure
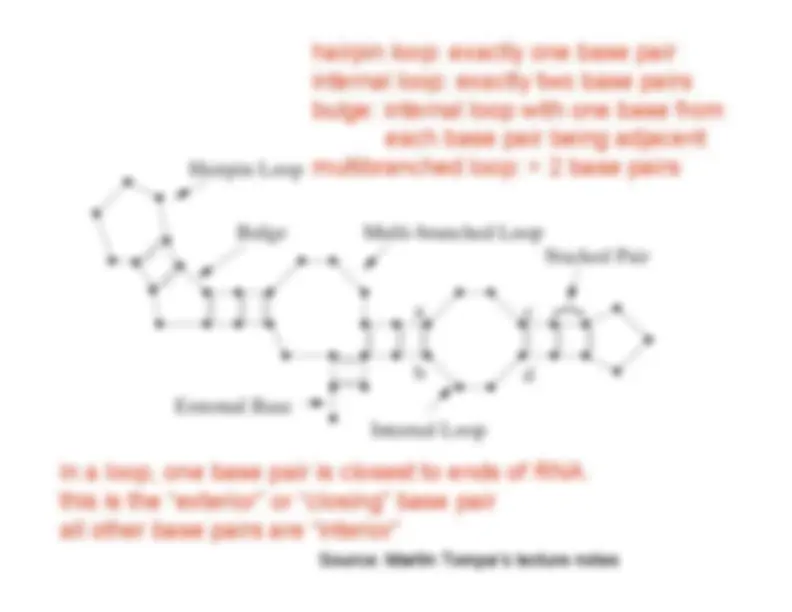
Secondary structure elements
Loop: single stranded subsequences bounded by base pairs loop at the end of a stem stem loop single stranded bases within a stem … only on one side of stem … on both sides of stem
Non-canonical base pairs
- G-C and A-U are the canonical base pairs
- G-U is also possible, almost as stable
Pseudoknot
2 11 9 18 (9, 18) (2, 11) NOT NESTED
Pseudoknot problems
- Pseudoknots are not handled by the algorithms we shall see
- Pseudoknots do occur in many important RNAs
- But the total number of pseudoknotted base pairs is typically relatively small