Download Pharmaceuticals analytical Chemistry and more Lecture notes Analytical Chemistry in PDF only on Docsity!
- What is the definition of SOP? SOPs are detailed written instructions for the operations routinely performed in the course of any activities associated with pharmaceutical manufacturing. Or A written authorized procedure which gives instructions for performing operations not necessarily specific to a given product / material, but of a more general nature the equipments preventive maintenance and cleaning; recall of products; purchasing; cleaning of premises and environmental control; sampling and inspection etc. Or These are guidelines which describe how the activity is to be performed. To achieve uniformity of results by each individual, it is mandatory to follow these guidelines. SOP is like a “TELL and SHOW” concept. Tell – means to establish and teach how the activity is to be carried out. Show – means to provide the documented proof for the activity carried out.
- What are the contents of the SOP? Objective/Purpose, Scope, Responsibility, Accountability, Procedure, List of formats/Annexure, Abbreviations, Reference, Revision History
- Which information should master document carry on every page not just one of the pages to meet GMP? Page number, document reference number and authorizing signatures
- How many SOPs required for equipment and what are those? Operation, Cleaning, Preventive maintenance/ Calibration, Sampling procedure
- What is the Batch production and control record (BPCR)? BPCR are prepared for each intermediate and API and include the complete information relating to the completion of each significant step in the Batch production.
- What is the Master production & control record (MPCR)? To ensure the uniformity from batch to batch, master production instructions for each intermediate and API are prepared, dated and signed by one person, immediately checked, dated and signed by a person in the quality unit.
- What are the content of the MPCR? The name of the intermediate or API being manufactured and an identifying document
reference code, if applicable A complete list of raw materials and intermediates designated by names or codes sufficiently specific to identify any special quality characteristics. An accurate statement of the quantity or ratio of each raw material or intermediate to be used, including the unit of measure. Where the quantity is not fixed, the calculation for each batch size or rate of production should be included. Variations to quantities should be included where they are justified The production location and major production equipment to be used Detailed production instructions, including the:
- Sequences to be followed
- ranges of process parameters to be used
- sampling instructions and in-process controls with their acceptance criteria, where appropriate
- time limits for completion of individual processing steps and/or the total process, where appropriate
- expected yield ranges at appropriate phases of processing or time -Where appropriate, special notations and precautions to be followed, or cross- references to these The instructions for storage of the intermediate or API to ensure its suitability for use, including the labeling and packaging materials and special storage conditions with time limits, where appropriate.
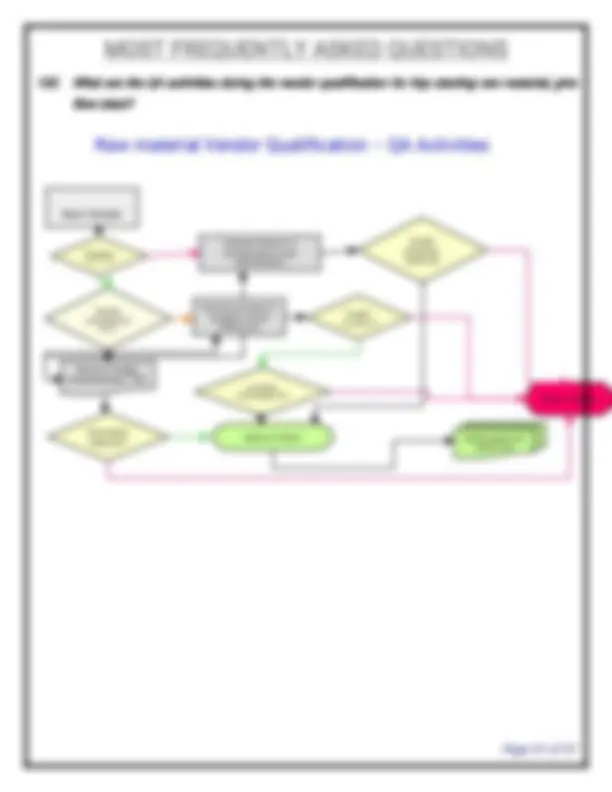
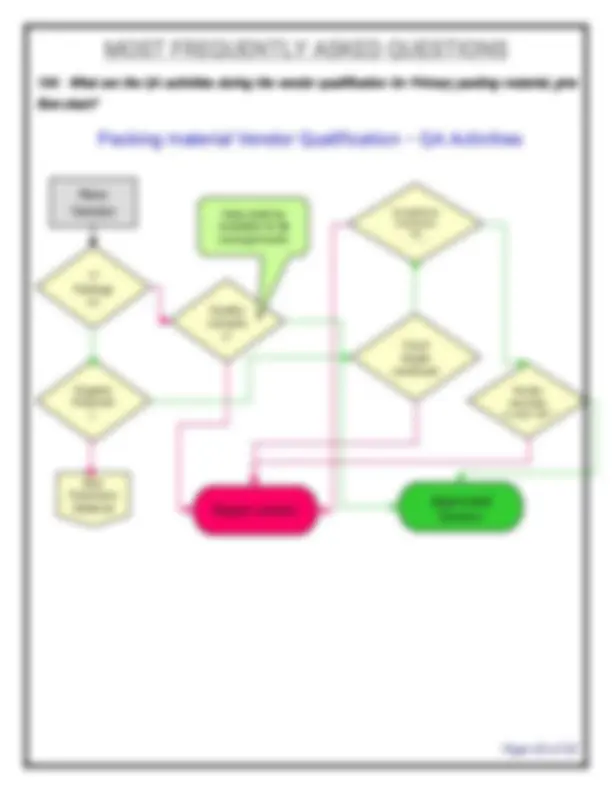
- What is the list SOPs required in QA department? SOP for SOP, SOP for format preparation, change control, deviation, Non-conformance products, market complaints, product recall, returned goods, vendor qualification, preparation of BPCR & MPCR, Assigning of Mfg. date & Expiry date, annual product review, corrective action & preventive action, process validation, cleaning validation, equipment qualification, glossary of terms, document control, Review of BPCR & analytical test report, batch numbering system, labeling practice, personnel training, BPCR issue and retrieval, batch release, self inspection (internal audit), file numbering system, preparation of organo-gram, preparation of COA, specimen signatures, Reprocess & rework of intermediates / API, Job responsibilities, Technology transfer, measurable quality objectives etc.
Australia (AS 1386): 0.035, 0.35, 3.5, 35, 350, 3500 Germany (VDI 2083): 1, 2, 3, 4, 5, 6
- What is the difference between GMP & cGMP? GMP: GMP is the part of Quality assurance which ensures that products are consistently produced and controlled to the quality standards appropriate to their intended use and as required by the marketing authorization. GMP are aimed primarily at diminishing the risks inherent in any pharmaceutical production. Such risks are essentially of two types:
- Cross-contamination (in particular of unexpected contamination)
- Mix-ups (confusion) cGMP: Current Good Manufacturing Practices. This means any procedure / system adopted by the manufacturer which proves to be necessary and important for identity, strength and purity of a product.
- What is the difference between Qualification and Validation? Qualification is equipment / instrument oriented but validation is process oriented.
- What is the definition of Validation? Validation is the documented program that provides a high degree of assurance that a specific process, method or system will consistently produce a result meeting predetermined acceptance criteria.
- What is the definition of Qualification? Qualification is the action of proving and documenting that any equipment or ancillary systems are properly installed, work correctly, actually leads the expected results. Qualification is part of validation, but the individual qualification steps alone do not constitute process validation.
- What are the types of validation? Process validation, Analytical method validation, cleaning validation, facility validation, Utility validation & software validation
- Definition of process validation and types of process validation? Process validation is the documented evidence that the process, operated within established parameters, can perform effectively and reproducibly to produce an intermediate / API meeting
its pre-determined specifications and quality attributes. Process validation is three types:
- Prospective process validation
- Concurrent process validation
- Retrospective process validation
- What is the prospective, concurrent and retrospective validation? Prospective process validation: Prospective Process validation shall be carried out for all the intermediate stages and Active Pharmaceutical Ingredients prior to the distribution of a new product. [ICH: GMP, EU: GMP, PIC/S: GMP] Concurrent process validation: Any validated process undergoes a change either for the equipment or addition, deletion of a critical manufacturing process step, scale up or scale down, the same needs to be validated concurrently. The validation is carried out only after a change of an existing validated process to support the change made or involve with the requirements. Or A subset of prospective validation in which API batches are released for distribution, based on extensive testing, before completion of process validation. Once data from additional batches produced under replicated conditions show uniformity, the process may be considered validated Or Concurrent validation can be conducted when data from replicate production runs are unavailable because only a limited number of API batches have been produced, API batches are produced infrequently, or API batches are produced by a validated process that has been modified. [ICH: GMP, EU: GMP, PIC/S: GMP] Retrospective process validation: Validation of a process for a product already in distribution based upon accumulated production, testing and control data. [ICH: GMP, EU: GMP, PIC/S: GMP]
- What do you mean by validation protocol and its contents of process validation? A written plan stating, how validation will be conducted and defining acceptance criteria
- What is the master document? Master document is a formally authorized source document relating to specifications, and / or manufacturing / analytical methods, which is protected from un-authorized access or amendment. Documents required describing the quality system requirements in the organization. Documents required describing the process or product characteristics. Documents required by various regulatory agencies as part of compliance to GMP requirements. Documents required for legal/ regulatory supports of the organization to meet the local regulations. Any other documents required by government / regulatory agency.
- What is documentation? All the written production procedures, instructions and records, quality control procedures and recorded test results involved in the manufacturing of a medicinal product.
- What is the Technology Transfer? In the pharmaceutical industry, “technology transfer” refers to the processes that are needed for successful progress from drug discovery to product development to clinical trials to full- scale commercialization or it is the process by which a developer of technology makes its technology available to commercial partner that will exploit the technology. To assure the drug quality, it is desire to make sure 5 W’s and 1 H, that is what 1 , when 2 , and why 3 information should be transferred to where 4 and by whom 5 and how to transfer, then share knowledge and information of the technology transfer each other between stake holders related to drug manufacturing.
- What are the names of different countries of GMP guidelines for manufacturing of API? WHO GMP - Geneva ICH Q7A – Europe, Japan & US EU GMP - Europe MCC – South Africa APIC GMP – Active Pharmaceutical Ingredient Committee (A sector group of CEFIC)
USFDA GMP – United States of America PIC/S GMP- Germany Schedule M – Indian
- What is preventive maintenance? It is periodic inspection and minor repairs of equipment as per schedule given in the SOP. This enables smooth operation and long life of the equipment. It also avoids major breakdown of the equipment during manufacturing of the product. There are two types of maintenance. Preventive maintenance: Schedule maintenance before any break down of machinery which prevents the machine break down. Breakdown maintenance: Maintenance was done after stopping machine breakdown. Weekly, Monthly, Quarterly, Half yearly and Yearly preventive maintenance
- What do you mean by “Quality Assurance”? The sum total of the organized arrangements made with the objects of ensuring that all APIs are of the quality required for their intended use and the quality systems are maintained.
- What are the types of different training programs?
- Induction training
- Job oriented training
- cGMP training
- On-going training
- What is cGMP? Current Good Manufacturing Practices. This means any procedure / system adopted by the manufacturer which proves to be necessary and important for identity, strength and purity of a product.
- What are the requirements for the equipment used in the manufacturing of process of API? Material of construction used for equipment should not React with component Get corroded, cause rusting Impart any impurities, absord
- Composition / Information on Ingredients
- Hazards identification
- First Aid measures
- Fire fighting measures
- Accidental release measures
- Handling & storage
- Exposure controls / Personal protection
- Physical & Chemical properties
- Stability & Reactivity
- Toxicological information
- Ecological information
- Disposal consideration
- Transport information
- Regulatory information
- Other information
- What is the static electricity? Denoting / pertaining to electricity which is at rest. The electricity which is present on surface of a non-conductive body, where it is trapped from escaping, is called static electricity.
- What is the different types of Qualifications and write its flow? Qualifications are as follows: Design Qualification, Installation Qualification, Operational Qualification, and Performance Qualification. URS/DS -----FAT-----SAT-----DQ-----IQ-----OQ-----PQ
- What is audit/inspection and Why quality audit? Write different types of audits/inspection? A planned and systematic examination and check of a system, procedure or operation in order to monitor compliance with and the effectiveness of established standards and to allow for improvement and corrective measures where required. Quality audit because of: To assess the effectiveness of the quality management system Assessing conformance
Investigating problems Continual improvement of performance Assessing for Registration Reducing cost of operation Legal requirement Types: 1. Study/test based inspection
- Facility based inspection
- Process based inspection
- Why nitrogen gas used in the manufacturing area at room temperature and why not other gas? Because of nitrogen is chemically less reactive and does not react with other elements at ordinary temperature. It is due to strong bonding in its molecule.
- What are the different types of cleanings? There are three types of cleanings: Batch to Batch cleaning Periodically cleaning Product change over cleaning
- What is blending? Blending is defined as the process of combining materials within the same specification to produce a homogeneous intermediate or API.
- What is expiry date & re-test date? Expiry date: The date place on the container / labels of an API designated the time during which the API is expected to remain within established shelf life specifications if stored under defined conditions and after which it should not be used. Re-test date: The date when a material should be re-examined to ensure that it is still suitable for use. The period of time during which the drug substance is expected to remain within its specifications and therefore, can be used in the manufacturing of the drug product, provided that drug substance has been stored under the defined conditions.
- What is difference between reprocess & rework? Reprocess: Introducing an intermediate or API, including one that does not conform to
Level 2 (Major): Are those that are likely to have a significant impact on the
quality attributes of the product.
The type of reasons for change control:
- Regulatory requirement
- GMP implementation / enhancement
- Quality improvement
- Capacity enhancement
- Introduction of new product in existing facility
- Cost reduction
- Automation
- Aging of facility
- To manage the unavoidable situation
- Market requirement
- What is contamination and cross-contamination? Contamination: The undesired introduction of impurities of a chemical or Microbiological nature, or of foreign matter, in to or onto a raw material, intermediate, or API during production, sampling, packaging or repackaging, storage or transport. Cross-contamination: Contamination of a material or of a product with another material or product.
- What is Batch number and batch? Batch Number: A unique combination of numbers, letters, and/or symbols which identifies a batch (or lot) and from which the production and distribution history can be determined Batch: A specific quantity of material produced in a process or series of processes so that it is expected to be homogeneous within specified limits. In the case of continuous production, a batch may correspond to a defined fraction of the production. Batch size may be defined either by a fixed quantity or the amount produced in a fixed time interval.
- What is quarantine? The status of materials isolated physically or by other effective means pending a decision on their subsequent approval or rejection.
- What is definition of critical process parameters? A process parameter whose variability has an impact on a critical quality attribute and
therefore should be monitored or controlled to ensure the process produces the desired quality.
- What is mother liquor? The residual liquid which remains after the crystallization or isolation processes. Mother liquor may contain un-reacted materials, intermediates, levels of the API and/or impurities. It may be used for further processing.
- What is the difference between theoretical and expected yield? Theoretical yield: The quantity that would be produced at any appropriate phase of production, based upon the quantity of material to be used, in the absence of any loss or error in actual production. Expected yield: The quantity of material or the percentage of theoretical yield anticipated at any appropriate phase of production based on previous laboratory, pilot scale, or manufacturing data.
- What is OOS? Out of Specification (OOS) results are those results, generated during testing that do not comply with the relevant specification or standards or with the defined acceptance criteria.
- What is CAPA? CAPA is the Corrective Action & Preventive Action. Corrective Action: Action taken to eliminate the causes of an existing non-conformity, defect or other undesirable situation to prevent recurrence. [Actions taken after the occurrence of a defect or problem to stop the same from recurrence]. Preventive Action: Action taken to eliminate the causes of potential non-conformity, defect or other undesirable situation to prevent occurrence. [Actions initiated before the occurrence of a defect or problem to prevent the same occurrence].
- What is the ICH? Write its aim/purpose and names of the different parties & different regions? ICH means “International conference on harmonization”. Aim/Purpose: “Ensure good quality, safety and effective medicines are developed and registered in the most effective manner, through harmonization of technical requirements” Different Parties:
- European commission – European Union (EMEA)
route in a heat exchanges? The following characteristic of the fluid are to be considered while deciding its route in a heat exchanger: a) Viscosity b) Fouling c) Corrosiveness d) Pressure
- When steam distillation recommended? a) To separate appreciable quantities of higher boiling materials. b) To separate relatively small amounts of volatile impurity from a large amount of material. c) Where use of direct-fired heaters is detrimental to the materials. d) Where the material is to be subjected to distillation is thermally unstable or will react with other component associated with it at the boiling temperature. e) Where the material cannot be distilled by in-direct heating even under low pressure because of the high boiling temperature.
- What is the difference between instrument & equipment? Instrument: A device that takes a physical measurement and displays a value or has no control or analytical functions. e.g.: Stop watch, timers & thermometer. [A device <chemical, electrical, hydraulic, magnetic, mechanical, optical, pneumatic> used to test, observe, measure, monitor, alter, generate, record, calibrate, manage or control physical properties, movements, or other characteristics]. Equipment: A device or collection of components that perform a process to produce a result. [The collective analytical measurement instruments in conjunction with firmware, assembled to perform a mechanical process]
- What is HVAC? The HVAC is designed to circulate the air in the area after passing it over cooling & heating coils to maintain the required environmental conditions & passing it through the series of filters to maintain desired cleanliness level in the area. The air in-take and out-take of the system is designed to maintain certain degree of pressure gradient in the area as per requirements. Or HVAC system function is to condition (heating & cooling), replace (makeup, fresh air, oxygen replacement), and pressurize (contaminant) and clean (filter) the air in the environment to meet the required operational conditions.
To achieve this objective, electrical, mechanical & electronic components are arranged in several configurations such that they produce the expected results.
- What is the meaning of Q, S, E, M in the ICH? “Q” stands for Quality; “S” stands for Safety, “E” stands for Efficacy and “M” stands for Multi dispensary
- How many guidelines are present in Q & what are those, describe in detail? In Quality (Q), total 10 guidelines are present. Those are as follows:
- Q1 - Stability
- Q2 - Analytical Method validation
- Q3 - Impurities
- Q4 - Pharmacopoeia
- Q5 - Biotechnological quality
- Q6 - Specification
- Q7 - Good Manufacturing Practice (GMP)
- Q8 - Pharmaceutical Development
- Q9 - Quality Risk Management
- Q10 - Pharmaceutical Quality System
- How many types of raw material and packing material? Raw materials are classified into two types. Those are as follows:
- Key raw material
- Other raw material Packing materials are classified into two types. Those are as follows:
- Primary Packing material
- Secondary Packing material
- Define the Key raw material/ starting material & primary packing material? Key raw material/starting material: Starting material shall be defined as that which is Incorporated as a significant structural fragment of the API / Drug Intermediate and Having significant effect on the Quality and Yield of the product. Starting material shall be identified in TDP. Primary Packing material: Packing material, which come in direct contact with the API/Intermediate are considered as Primary packing material.
- What are the contents in the BPCR? BPCR contains the following contents, but not limited:
- Product Name 2. Stage
- BPCR Document Number 4. MPCR Reference Number
- Batch Number 6. Date of Manufacturing
- Date of Expiry/Re-test 8. Batch release details
- List of equipments used 10. List of raw materials & Quantity with UOM
- General instructions, Control & Safety instructions
- Detailed step wise written manufacturing procedures
- Actual results record for critical process parameters
- Identity of In-process & Laboratory test results
- Signatures of person performing details along with supervising details
- Description of Packaging details
- Yield calculation
- Representative of labels for intermediates / raw materials
- Deviation details
- Batch starting & completion date
- What is OOT and define? “OOT” stands for Out Of Trend. It means any test results obtained for a particular batch that is markedly different the results of the batches in a series obtained using a same validated method.
- How will you prevent cross-contamination between two different products manufactured in the one production block? By maintaining the proper pressure differential between the rooms with two Air handling units (if re-circulation) / one Air handling unit (if 100% fresh air)
- What is limit of Temperature and relative humidity in the pharma area? Temperature: 25±2˚C & Relative Humidity: 50±5%
- What is the difference between dedicated and non-dedicated equipments? Dedicated equipment: It is used solely for the production of a single product or product line. Concerns over cross-contamination with other products are markedly reduced. Dedicated equipments must be clearly identified with the restrictions of use in order to prevent potential
errors during cleaning and preparation. Non-dedicated equipment: Where the same piece of equipment is utilized for a range of products formulations. The prevent of cross-contamination between products becomes the main objective in the cleaning validation effort. Clearly, cleaning non-dedicated equipments represents a more significant obstacle to overcome.
- Which instrument is used for the measuring of RPM? Techo meter is used for the measurement of RPM.
- Why three batches consider for the validation? Because of First one is for information, Second one is for confirmation and Third one is for evidence.
- If one batch is failed during the validation, then what will you do for completion of validation? When a quality parameter fails with respect to the specification, a deviation report shall be raised and the investigation shall be conducted immediately for the identification of failure. If the reason for failure is identified, one more consecutive batch shall be considered for the validation run by taking preventive actions to avoid those failures (If necessary revise the MPCR and BPCR). If the reason is unidentified, another three consecutive batches shall be taken for validation
- What are specifications of Purified water as per any pharmacopoeia? Tests (^) Ph. Eur. Description Clear, colorless liquid Acidity /Alkalinity The solution is not colored red/The solution is not colored blue. Oxidisable substances The solution remains faintly pink Chlorides The solution shows no change in appearance for at least 15 min Sulphates The solution shows no change in appearance for at least 1 hour Ammonium Maximum 0.2 ppm. Calcium and magnesium A pure blue colour is produced. Residue on evaporation Maximum 0.001 per cent Aluminum Maximum 10 ppb, Nitrates NMT 0.2 ppm