Download Understanding Crystal Structures: Close-Packed Atoms & Defects in FCC and HCP and more Lecture notes Materials science in PDF only on Docsity!
2. 0 STRUCTURE OF METALS AND ALLOYS
Since the electrons in a metallic lattice are in a “gas,” we must use the core electrons and nuclei to determine the structure in metals. This will be true of most solids we will describe, regardless of the type of bonding, since the electrons occupy such a small volume compared to the nucleus. For ease of visualization, we consider the atomic cores to be hard spheres. Because the electrons are delocalized, there is little in the way of electronic hindrance to restrict the number of neighbors a metallic atom may have. As a result, the atoms tend to pack in a close-packed arrangement, or one in which the maximum number of nearest neighbors (atoms directly in contact) is satisfied. Refer to Figure 2.1. The most hard spheres one can place in the plane around a central sphere is six, regardless of the size of the spheres (remember that all of the spheres are the same size). You can then place three spheres in contact with the central sphere both above and below the plane containing the central sphere. This results in a total of 12 nearest-neighbor spheres in contact with the central sphere in the close-packed structure. Figure 2.1 Close-packing of spheres. (a) Top view, (b) side view of ABA structure, (c) side view of ABC structure Closer inspection of Figure 2.1 (a) shows that there are two different ways to place the three nearest neighbors above the original plane of hard spheres. They can be directly aligned with the layer below in an ABA type of structure, or they can be rotated so that the top layer does not
align core centers with the bottom layer, resulting in an ABC structure. This leads to two different types of close-packed structures. The ABAB_..._ structure (Figure 2.1 b) is called hexagonal close-packed (HCP) and the ABCABC_..._ structure is called face-centered cubic (FCC). Remember that both of these close- packed arrangements have a coordination number (number of nearest neighbors surrounding an atom) of 12: 6 in plane, 3 above, and 3 below. Figure 2.2 The extended unit cell of the hexagonal close-packed (HCP) structure Keep in mind that for close-packed structures, the atoms touch each other in all directions, and all nearest neighbors are equivalent. Let us first examine the HCP structure. Figure 2.2 is a section of the HCP lattice, from which you should be able to see both hexagons formed at the top and bottom of what is called the unit cell. You should also be able to identify the ABA layered structure in the HCP unit cell of Figure 2.2 through comparison with Figure 2.1. Let us count the
three atoms along a diagonal. These three atoms form the diagonal on the face of the FCC unit cell, which is shown in Figure 2.3. There is a trade-off in doing this: It is now difficult to see the close-packed layers in the FCC structure, but it is much easier to see the cubic structure (note that all the edges of the faces have the same length), and it is easier to count the total number of atoms in the FCC cell. In a manner similar to counting atoms in the HCP cell, we see that there are zero atoms completely enclosed by the FCC unit cell, six face atoms that are each shared with an adjacent unit cell, and eight corner atoms at the intersection of eight unit cells to give Remember that both HCP and FCC are close-packed structures and that each has a coordination number of 12, but that their respective unit cells contain 6 and 4 total atoms. We will now see how these two special close-packed structures fit into a larger assembly of crystal systems. 2.1 Crystal Structures Our description of atomic packing leads naturally into crystal structures. While some of the simpler structures are used by metals, these structures can be employed by heteronuclear structures, as well. We have already discussed FCC and HCP, but there are 12 other types of crystal structures, for a total of 14 space lattices or Bravais lattices. These 14 space lattices belong to more general classifications called crystal systems , of which there are seven.
Figure 2.4 Definition of a coordinate system for crystal structures. Before describing each of the space lattices, we need to define a coordinate system. The easiest coordinate system to use depends upon which crystal system we are looking at. In other words, the coordinate axes are not necessarily orthogonal and are defined by the unit cell. This may seem a bit confusing, but it simplifies the description of cell parameters for those systems that do not have crystal faces at right angles to one another. Refer to Figure 2.4. For each crystal system, we will define the space lattice in terms of three axes, x, y , and z , with interaxial angles α, β, γ. Note that the interaxial angle α is defined by the angle formed between axes z and y , and also note that angles β and γ are defined similarly. Only in special circumstances are α, β, γ equal to 90 ◦. The distance along the y axis from the origin to the edge of the unit cell is called the lattice translation vector , b. Lattice translation vectors a and c are defined similarly along the axes x and z , respectively. The magnitudes (lengths) of the lattice translation vectors are called the lattice parameters , a , b , and c. We will now examine each of the seven crystal systems in detail. 2.1.1 Crystal Systems. The cubic crystal system is composed of three space lattices, or unit cells, one of which we have already studied: simple cubic (SC), bodycentered cubic (BCC), and
Figure 2.5 Summary of the 14 Bravais space lattices.
Figure 2.6 A tetrahedral interstice. Figure 2.7 An octahedral interstice.
There is only one space lattice in the rhombohedral crystal system. This crystal is sometimes called hexagonal R or trigonal R , so don’t confuse it with the other two similarly-named crystal systems. The rhombohedral crystal has uniform lattice parameters in all directions and has equivalent interaxial angles, but the angles are nonorthogonal and are less than 120O. The crystal descriptions become increasingly more complex as we move to the monoclinic system. Here all lattice parameters are different, and only two of the interaxial angles are orthogonal. The third angle is not 90◦. There are two types of monoclinic space lattices: simple monoclinic and base-centered monoclinic. The triclinic crystal, of which there is only one type, has three different lattice parameters, and none of its interaxial angles are orthogonal, though they are all equal. Finally, we revisit the hexagonal system in order to provide some additional details. The lattice parameter and interaxial angle conditions shown in Figure 2.5 for the hexagonal cell refer to what is called the primitive cell for the hexagonal crystal, which can be seen in the front quadrant of the extended cell in Figure 2.2. The primitive hexagonal cell has lattice points only at its corners and has one atom in the center of the primitive cell, for a basis of two atoms. A basis is a unit assembly of atoms identical in composition, arrangement, and orientation that is placed in a regular manner on the lattice to form a space lattice. You should be able to recognize that there are three equivalent primitive cells in the extended HCP structure. The HCP extended cell , which is more often used to represent the hexagonal structure, contains a total of six atoms, as we calculated earlier. In the extended structure, the ratio of the height of the cell to its base, c/a , is called the axial ratio. 2.1.2 Defects Now that the most important aspects of perfect crystals have been described, it is time to recognize that things are not always perfect, even in the world of space lattices. This is not necessarily a bad thing. As we will see, many important materials phenomena that are based on defective structures can be exploited for very important uses. These defects , also known as imperfections , are grouped according to spatial extent. Point defects have zero dimension; line defects , also known as dislocations , are one dimensional; and planar defects such as surface defects and grain boundary defects have two dimensions. These defects may occur individually or in combination.
Let us first examine what happens to a crystal when we remove, add, or displace an atom in the lattice. We will then describe how a different atom, called an impurity (regardless of whether or not it is beneficial), can fit into an established lattice. When an atom is missing from a lattice, the resulting space is called a vacancy (not to be confused with a “hole,” which has an electronic connotation), as in Figure 2.9. Figure 2.9 Representation of a vacancy and self-interstitial in a crystalline solid.
Figure 3.1 Representation of Frenkel and Schottky defects in a crystalline solid 2.1.3 Line Defects and Dislocations We now move on to defects that have some spacial extent, even if only in one dimension. As we continue to increase the geometric complexity of these defects, you may find it more difficult to visualize them. As with crystal structures, three-dimensional models may help you with visualization, and do not limit yourself to one representation of a specific defect—look for multiple views of the same thing. The first type of one-dimensional defect, or line defect , is called a dislocation. A dislocation is a linear disturbance of the atomic arrangement in a crystal caused by the displacement of one group of atoms from an adjacent group. There are three types of dislocations: edge dislocations, screw dislocations , and a combination of these two, termed mixed dislocations. An edge dislocation occurs when a single atomic plane does not extend completely through the lattice. The termination of this half-plane of atoms creates a defect line (dislocation line) in the lattice (line DC in Figure 3.2). The edge dislocation is designated by a perpendicular sign, either ⊥ if the plane is above the dislocation line or _ if the plane is below the dislocation line. Edge dislocations can be quantified using a vector called the Burger’s vector , b , which represents the
relative atomic displacement in the lattice due to the dislocation (see Figure 3.3). The Burger’s vector is determined as follows: Define a positive direction along the dislocation line. This is usually done into the crystal. Construct a plane of atoms perpendicular to the dislocation line. Trace out a clockwise path around the dislocation line moving n lattice vectors in each of the four mutually perpendicular directions. The Burger’s vector is drawn from the finish to the start of the path. Figure 3.2 Edge dislocation DC results from introducing an extra half-plane of atoms ABCD , where DC is considered a positive dislocation designated by ⊥.
To this point, we have concentrated on single crystals. Most crystalline materials are polycrystalline —that is, composed of many small crystals, or grains , that usually have random crystallographic orientation relative to each other. Unless the grains are at a surface, they are adjacent to other grains, not necessarily of the same orientation. The region where they intersect is called a grain boundary. There are two general types of grain boundaries: tilt and twist. A tilt grain boundary is actually a set of edge dislocations (see Figure 3.4). The angle of misorientation , θ , characterizes the tilt grain boundary and is defined as the angle between the same directions in adjacent crystals. Figure 3.4 Representation of a tilt grain boundary.
2.2 STRUCTURE OF CERAMICS AND GLASSES
Inorganic materials constitute the largest class of solids in the world. We have already described metals; and while they are not organic (they contain no biological carbon), they are also not inorganic in the strict sense of the word—they are metals due to the unique characteristics of their valence electronic structure. Inorganic materials are typically compounds, such as metal oxides, carbides, or nitrides. They possess many interesting properties that we will only begin to describe at this point. They can also differ structurally from other types of materials like metals and polymers. Let us begin by describing the structure of inorganic materials. 2 .2.1 Pauling’s Rules Recall that the structure of a crystal is determined mostly by how the atoms pack together. The same is true of binary compounds such as alloys, and of binary compounds that contain noncovalent bonds, such as ionic compounds. In addition to the concept of electronegativity, Linus Pauling also produced a set of generalizations that are used to describe the majority of ionic crystal structures. Pauling’s first rule states that coordination polyhedra are formed. Coordination polyhedra are three-dimensional geometric constructions such as tetrahedra and octahedra. Which polyhedron will form is related to the radii of the anions and cations in the compound. Consider the two dimensional representation of a binary ionic compound shown in Figure 3.5. The anions (open circles) are larger than the cations, and a central cation cannot remain in contact with the surrounding anions if the anion radius is larger than a certain value. Thus, the structure in Figure 3.5 (c) is unstable, whereas the structures in Figures 3.5(a) and 3. (b) are both stable. Note that the cation–anion distance is simply the sum of the cation–anion radii for the stable structures. It is also true that the coordination number is determined by the radius ratio of the two ions (Ranion/Rcation ). The larger the central cation, the more anions that can be packed around it. For each coordination number, there is some critical value of the radius ratio above which the structure will not be stable. Pauling’s second rule on the packing of ions states that local electrical neutrality is maintained. We use a quantity called bond strength to assure electrical neutrality, where the bond strength is the ratio of formal charge on a cation to its coordination number. For example, silicon has a formal charge of +4 and a coordination number of 4, so that its strength is 4 / 4 = 1. For aluminum, a formal charge of +3 and a coordination number of six gives a bond strength of 1/2. So, the total strength of the bonds reaching an anion from all surrounding cations is equal to the charge of the anion. Pauling’s third rule tells us how
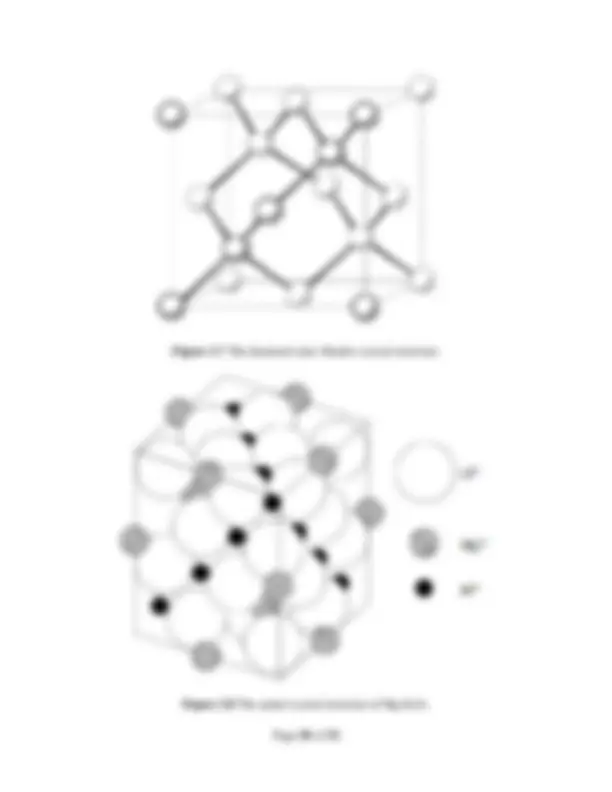
Figure 3.6(a) Tetrahedra ( top ) and octahedra ( bottom ) linked by sharing (a) corners, (b) edges, and (c) faces. Figure 3.6(b) The rock salt crystal structure. 2 .2.2.2 Diamond Structure. Another common ceramic structure arises when the tetrahedral sites in an FCC array of anions are occupied. For example, an FCC array
Figure 3.7 The diamond (zinc blende) crystal structure. Figure 3.8 The spinel crystal structure of MgAl 2 O 4.