Scarica Allineamento di Sequenze Biologiche: Metodi e Applicazioni e più Appunti in PDF di Bioinformatica solo su Docsity!
BIOINFORMATICA AVANZATA ANNO 2020/
PRIMA LEZIONE 1/10/
ANALISI DI SEQUENZA
Tramite la biologia evolutiva, si è visto che se confrontiamo due sequenze (DNA, RNA, proteina) di due specie diverse, se queste sono simili molto probabilmente svolgeranno la stessa funzione, molto probabilmente queste due specie hanno degli antenati in comune. Identitá : confronto residui identici di due sequenze o percentuale di molecole che corrispondono nell’allineamento tra due sequenze. SEQUENZA A= 320 amminoacidi e SEQUENZA B= 450 amminoacidi Tra queste due sequenze sono stati trovati 100 amminoacidi in comune I (identitá) è calcolata come il rapporto tra il numero di amminoacidi in comune e la sequenza piú corta = 100/ 320= 31.25% Similaritá: amminoacidi diversi ma con caratteristiche simili che se vengono sostituiti tra di loro non danno alterazioni. Se tra le sequenze precedenti, vi sono 23 amminoacidi simili, la S (similaritá) si calcola sommando gli amminoacidi identici a quelli simili e dividendo per la sequenza minore = 100+23/320=38.44% Queste due sequenze sono quindi identiche al 31% e simili al 38%. LCS= MISURA DI SIMILARITÁ se ho una stringa x formata da un tot di caratteri, la sottosequenza di x è formata dai caratteri estratti dalla stringa x nello stesso ordine ma non necessariamente vicina. Se la stringa ha una lunghezza n, la sottosequenza sará 2 n Se consideriamo S1 e S2 queste potrebbero avere sottosequenze in comune e quindi si fa riferimento a LCS che rappresenta la massima sottosequenza comune, cioè la sottosequenza comune ad entrambe che ha peró lunghezza massima. Questa puó non era unica, ve ne possono essere diverse ma che abbiano tutte la stessa lunghezza (massima); maggiore è la lunghezza di LCS maggiore sará la similaritá e quindi in grado di sovrapposizione. PROGRAMMAZIONE DINAMICA
È un modo per risolverei problemi procedendo per step, creando dei sottoproblemi che vengono risolti singolarmente, tutte le soluzioni si uniscono per risolvere il problema iniziale. Per le suddivisioni si considerano 2 residui alla volta, considerando due sequenze di lunghezza m e n si considera l’equazione di ricorrenza: se consideriamo la sequenza S ATACGGT di lunghezza m=7 e la sequenza T TATTCT di lunghezza n= si confrontano per primi i residui che si trovano in ultima posizione, in questo caso notiamo che entrambe le sequenze terminano con T, quindi sicuramente la T fa parte della LCS. LCS [ATACGGT , TATTCT] puó essere considerato anche come LCS [m , n ]
Come si prosegue: si considera la prima casella vuota, che sarebbe la casella LCS [1 , 1], si considerano le due basi, quindi in questo caso A e C, e si vanno a considerare le 3 celle numerate, quella in diagonale, quella superiore e quella inferiore. Se i due residui sono uguali, si considera la casella diagonale e si aggiunge +1 al numero della casella in diagonale. Se come nel nostro caso, i due residui sono diversi, allora si considerano le caselle in alto e in basso e si prende il numero maggiore tra i due, nel nostro caso in entrambe c’è il numero 0 quindi è indifferente quale casella si sceglie. Procedendo in questo modo si compila tutta la matrice, e il numero di nostro interesse è l’ultimo numero nell’ultima casella in basso a destra, quello ci indica la lunghezza di LCS, da quel numero si procede a ritroso in diagonale, considerando gli elementi che abbiamo preso con la casella in diagonale ed escludendo quelli presi con le caselle superiori e inferiori, per avere tutta la sottosequenza.
Questo procedimento puó essere svolto con exel: dove le righe sono numerate da 1 e non da 0 e le colonne sono rappresentate da lettere. In questo caso la prima casella sará C3 e la formula sará cosi cambiata. Questa formula puó essere scritta in exel usando la funzione SE e si va a verificare se una data condizione è vera o meno. Questa formula puó essere copiata da una cella all’altra, exel pero aggiorna l’indice delle righe e delle colonne, quindi bisogna inserire il simbolo del dollaro $ che blocca la riga e la colonna: SE C$1=$A alla fine per ricavare la sequenza posso colorare le celle selezionando la matrice, andando su formattazione condizionare, poi su regole evidenziazione celle, su altre regole e inserire la formula E (C$1=$A3) in modo tale che si evidenziano solo le celle in cui Sm=Tn. SECONDA LEZIONE 8/10/ DISTANZA DI EDITING È una misura di similaritá che serve per misurare il numero di operazioni che servono per poter trasformare una stringa in un’altra, quindi è una misura della differenza tra le due stringhe, maggiore è la differenza e minore sará la similaritá. Le operazioni di editing sono 3:
- Inserimento di un nuovo elemento;
- Cancellazione di un elemento;
- Sostituzione di un elemento. Le prime due sono dette operazioni indel e vengo rappresentate da ‘’-‘’. É sempre possibile trasformare una sequenza perché si potrebbero cancellare tutti gli elementi della prima sequenza e sostituirli con gli elementi della seconda. Esempio In questo caso si puó trasformare la prima stringa nella seconda, effettuando 3
Infine, si puó effettuare un inserimento della C nella prima stringa. Nella sostituzione avró [ m-1, n-1] perché ho effettuato un’operazione in entrambe le stringhe mentre nella cancellazione effettuiamo un’operazione solo nella prima stringa e non nella seconda, quindi si sottrae ad m mentre n rimane invariato. Infine con l’inserimento, io sottraggo il residui alla seconda stringa quindi avró n-1 ma nella prima io vado ad inserire una stringa quindi rimane m. Le prime due possono essere rappresentate da un’unica equazione considerando delta che se è uguale a 0 sta a significare che i due residui sono uguali e che quindi si ha corrispondenza, se delta è uguale a 1 sta a significare che i residui sono diversi e che quindi vi sará una sostituzione. Anche in questo caso si costruisce una matrice, ma qui i prima valori non saranno tutti 0 ma andranno dallo 0 in poi. Considerando la prima cella [1,1], vediamo come vi sono due residui diversi (A e T) e quindi delta sará uguale a 1 e [m-1] [n-1] + delta rappresenta la cella in diagonale; [m-1, n] + 1 rappresenta la cella in alto; [m, n-1] + 1 rappresenta la cella di lato. Sostituendo avremo che: [m-1] [n-1] + delta= 0+1= 1 (perché nella cella in diagonale c’è 0 e delta è 1)
[m-1, n] + 1= 1+1= 2 (perché nella cella in alto c’è 1) [m, n-1] + 1= 1+1= 2 (perché nella cella di lato c’è 1) Di questi tre valori si prende solo il valore minore e si aggiunge alla casella. Si continua con lo stesso metodo per tutte le celle, le frecce rappresentano la direzione del numero nella cella, cioè se il numero piú piccolo aggiunto poi alla cella, era quello in diagonale, in alto o di lato. Se la freccia è in diagonale vuol dire che è stata effettuata una sostituzione, se è verso l’alto vuol dire che vi è stato un inserimento, se è di lato c’è stata una cancellazione. Quando vi è corrispondenza significa che i residui sono uguali quindi non si prendono in considerazione. L’ultimo numero della matrice rappresenta il numero di operazioni effettuate. Anche in questo caso, tutto ció puó essere effettuato su exel, utilizzando la funzione MIN che restituisce il valore minimo; la formula si traduce in: se C1 è uguale a A3 allora si effettua B2+0, altrimenti se sono diversi si effettua B2+1. Anche qui, si puó copiare la formula a tutte le celle inserendo il simbolo del dollaro, C$1=$A3 in modo da bloccare riga e colonna.
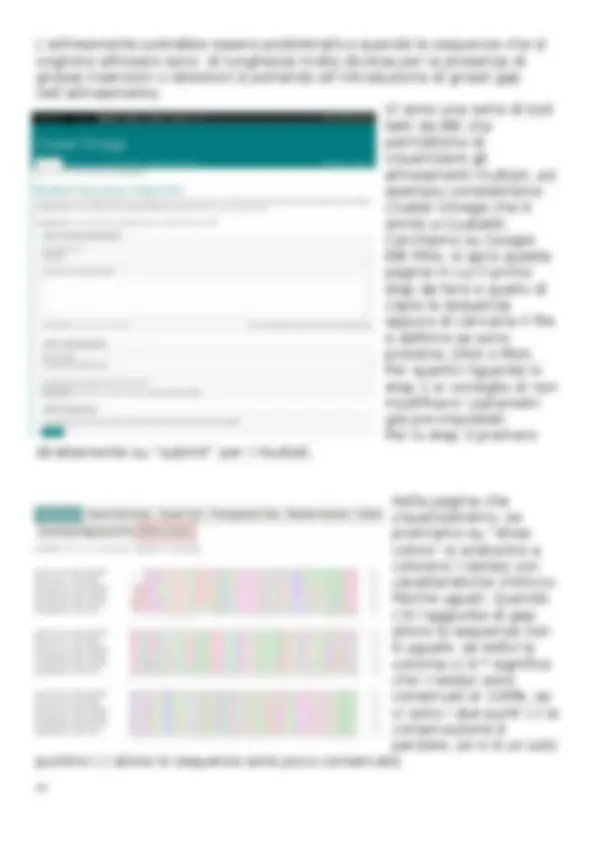
Primari: sono dei veri e propri archivi di struttura e sequenza dove tutti i dati non vengono rimossi ma ne vengono aggiunti sempre di nuovi. Secondari: possono integrare le informazioni di altri database e sono “curati” manualmente da teem di persone per raccogliere solo determinati tipi di informazioni. I vari database sono, quindi, collegati tra di loro e possono scambiarsi informazioni come si puó notare dallo schema che è una rappresentazione delle interazioni tra database diversi. Il principale database che useremo sará NCBI che è dell’istituto nazionale sanitario americano che si occupa di sviluppare software per analizzare i vari dati, e formato da un teem di figure con ruoli diversi; l’interesse principale è quello di sequenziare il genoma umano, ma ha avviati anche altri studi ad esempio sull’HIV. NCBI gestisce 39 database, ognuno focalizzato in un settore specifico: nucleotide e protein che permette di di scaricare nucleotidi e proteine, nucleotide database che permette di scaricare specifiche sequenze nucleotidiche, genebanck che è una banca dati primaria che colleziona sequenze biologiche dal 1992 con sequenze sottomesse da laboratori individuali, è una risorsa pubblica con consultazione gratis in cui si trovano sequenze di 250000 specie, il solo limite è la cosidetta ridondanza di informazione, in quanto si trovano dati relativi alla stessa sequenza che derivano da esperimenti e laboratori diversi. Poi abbiamo RefSeq che contiene informazioni su 60000 specie, è un database secondario quindi c’è uno staff che va ad integrare informazioni diversi sulle stesse sequenze dando cosí un solo risultato. Se lavoriamo su specie note come uomo o topo è meglio lavorare su refseq, al contrario se lavoriamo su specie meno note è meglio lavorare su Genebanck che viene costantemente aggiornato ogni due mesi. La prima cosa da fare su NCBI è quella di selezionare il database nella finestra in alto a sinistra, in questo caso abbiamo scelto di usare il database nucleotide, poi sulla barra di ricerca si
inserisce il gene da voler studiare e si preme search. A questo punto ci saranno una serie di risultati, possiamo quindi utilizzare queste tendine a destra e sinistra per inserire i cosidetti filtri, e quindi selezionare le informazione che vogliamo come ad esempio in alto a destra in “top organism” si puó selezionare la specie come homo sapiens, in alto a sinistra vi sono filtri come “molecular type” in cui possiamo ad esempio selezionare mRNA oppure “source database” in cui possiamo scegliere dove andare a cercare nello specifico i miei risultati come refseq. Una volta trovato il risultato che ci interessa, questa è la prima pagina che vediamo e la prima cosa che si va a notare è il cosidetto acession number che sarebbe il codice identificativo della scheda presa in esame, è un codice alfanumerico che il piú delle volte alla fine presenta un punto con un altro numero, quello indica gli aggiornamenti effettuati nel tempo. Possiamo avere altre informazioni principali come la lunghezza della sequenza (numero di bp), il tipo di molecola e la data dell’ultimo aggiornamento. Le parle chiavi, l’organismo da cui deriva, le referenze, gli autori dello studio, il giornale in cui è stato pubblicato l’esperimento. In alto sotto il titolo si ha la scritta FASTA, premendola si apre una nuova finestra dove tramite il tasto “send to” è possibile stampare la sequenza in un formato che prende il nome proprio di fasta che è equivalente a txt in quanto sono entrambi dei formati di testo. Scorrendo verso il basso si possono trovare altre informazioni piú specifiche e altri link che fanno aprire finestre in cui vi sono una serie di dati relativi al nostro gene, questi li vedremo nelle prossime lezioni.
refseq. A questo punto dei vari risultati andiamo a considerare quello con il numero di amminoacidi maggiore e prendere si il numero di amminoacidi che l’accession number. Andando verso il basso troviamo la sezione CDS e clicchiamo sul link corrispondente dove possiamo trovare le varie coordinate cromosomiche come il numero di cromosomi, la posizione di start e di and. QUARTA LEZIONE 15/10/ ALLINEAMENTO DI SEQUENZE BIOLOGICHE Si possono mettere a confronto le sequenze, sia di amminoacidi di DNA e RNA, ció permette di valutare la similaritá di due sequenze tenendo conto anche delle regioni in cui non vi è similaritá. L’allineamento si distingue in: Globale: confronta le sequenze (di lunghezza simile) nella loro interezza, includendo tutti i residui tre le due estremitá. Locale: identifica sottosequenze (di lunghezza differente) con maggior numero di residui uguali. Con gli allineamenti possiamo trovare i match che rappresentano i residui conservati (uguali), i mismatch che rappresentano una sostituzione o comunque la presenza di residui diversi, i gap che rappresentano l’inserzione o la delezione e in questo caso si inserisce un trattino orizzontale.
Gli allineamenti possono essere fatti in modo diverso, infatti date due sequenze di lunghezza n vi sono 2n^ modi di allineamenti diversi, per avere un allineamento migliore si assegna un punteggio ad ogni gap o sostituzione e alla fine si considera quello con score piú alto; Anche in questo caso possiamo ricorrere alla programmazione dinamica per l’allineamento globale tramite l’equazione di ricorrenza che prende il nome di needleman-wunsch in cui andremo ad avere uno score massimo, nel caso Sm=Tn avremo un match quindi residui uguali nel caso contrario avremo un mismatch.
Per quanto riguarda invece l’allineamento locale, l’algoritmo fu sviluppato da Smith e Waterman e dal calcolo del punteggio dell’allineamento va a scartare tutte le coppie che portano un punteggio negativo e restituisce la coppia di regioni il cui allineamento ha punteggio massimo. L’algoritmo è una variante di quello globale, con l’aggiunta di una quarta opzione, lo zero. Nella matrice che andremo a creare, anche in questo caso la prima riga e la prima colonna saranno tutti 0 altrimenti avremo tutti punteggi negativi.
Il procedimento rimane lo stesso dell’allineamento globale, si considerano i residui uguali e si utilizza il valore del match, per i residui diversi il valore del mismatch, nel caso in cui tutti e 3 i risultati sono negativi, si prende in considerazione lo 0. Alla fine, peró, non si considera il valore dell’ultima cella ma bensí il valore maggiore e si ritorna indietro fino ad arrivare allo 0. Per l’allineamento globale con BABA si setta NEED&WUNSCH, si completano la prima e la seconda stringa e si utilizza una penalitá con GAP a -1. Andando su score table, ci ritroviamo i nucleotidi in riga e colonna, e cosi nel caso di nucleotidi uguali si inserisce il valore di match, esempio 2, e nel caso di nucleotidi diversi si inserisce il valore di mismatch, esempio -2. Per l’allineamento locale il procedimento è il medesimo ma si utilizza SMITH&WATERM e si aggiunge lo 0 all’equazione. QUINTA LEZIONE 19/10/ MATRICI DI SOSTIRUZIONE L’allenamento si calcola come uno schema di punteggio generale di match e mismatch. Graficamente si rappresenta tramite delle matrici che
quindi per ricavare i valori di PAM2 di due proteine con due amminoacidi diversi tra loro possiamo effettuare PAM1xPAM1. Maggiore è il numero associato alle matrici e maggiore è la divergenza della sequenza. Con l’aumentare delle mutazioni, aumenta la probabilitá che singole mutazione revertano perché colpiscono siti giá mutati in precedenza. Quindi una matrice PAM80 non significa che vi sono proteine che divergono per l’80% ma solo del 50%, ma che sono avvenute 80 mutazioni ogni 100 amminoacidi. Se il numero associato aumenta, la divergenza aumenta in maniera esponenziale e poi si ferma. Gli amminoacidi con score alti con se stessi sono difficili da sostituire con altri come il triptofano W (11) e la cisteina C (9); le sostituzioni sono molto piú favorite tra amminoacidi che hanno caratteristiche simili. Matrici BLOSUM: (block substitution matrix) si basano su statistiche prese sul The blocks database che contengono allineamenti multipli tra regioni di amminoacidi di proteine omologhe. Vi sono una serie di blocchi che allineano sequenze che condividono una percentuale di identitá che va dal 30 al 95%. Ogni blocco è formato da sequenze continue senza gap; sono stati considerati circa 1500 blocchi di sequenze
amminoacidiche di circa 500 famiglie di proteine omologhe, per ogni blocco è stata calcolata la frequenza di ogni amminoacido e la probabilitá di sostituzione per ottenere uno score (misura) della sostituzione di un amminoacido con un altro, usando una formula ESEMPIO Consideriamo un blocco di 6 sequenze di lunghezza 4, con 24 amminoacidi totali. Le frequenze sono A viene contata 14 volte su 24, B viene contata 4 volte su 24 e infine C viene contata 6 volte su 24. A questo punto si considerano le sostituzione osservate: nel primo caso bisogna sostituire la A con la A, ovviamente solo le A che si trovano nella stessa colonna, ad esempio nella prima colonna troviamo 5 A, sostituiamo la prima A con le altre 4, la seconda con le altre 3, la terza con le altre 2 e la quarta con l’ultima, quindi nella prima colonna abbiamo 10 sostituzioni. Nella seconda colonna ci sono 6 A, sostituiamo la prima con le alte 5, la seconda con le altre 4, la terza con le altre 3, la quarta con le altre 2 e la quinta con l’ultima e avremo 15 sostituzioni. Nella terza colonna c’è una sola A che quindi non puó essere sostituita con le altre. E nella quarta colonna vi sono solo due A quindi è possibile 1 sola sostituzione. In totale avremo 10 sostituzioni dalla prima colonna, 15 dalla seconda e 1 dall’ultima, quindi vi sono 26 sostituzioni di A in A. Continuare cosí per tutte le sostituzioni possibili. Ora, invece dobbiamo ricavare le frequenze attese, nel primo caso di A sostituita con A dobbiamo moltiplicare le frequenze iniziali, quindi 14/24 x 14/24; mentre nel caso di A in B bisogna moltiplicare 14/24 x 4/24 ma dobbiamo ricordarci anche della sostituzione di B in A quindi avremo (14/21 x 4/24)
- (4/24 x 14/24); continuare cosí per tutte le sostituzioni.