Download Antibiotics (CELL WALL INHIBITORS) and more Study notes Pharmacology in PDF only on Docsity!
Cell Wall Inhibitors
Some antimicrobial drugs selectively interfere with synthesis of the bacterial cell wall—a structure that mammalian cells do not possess. The cell wall is composed of a polymer called peptidoglycan that consists of glycan units joined to each other by peptide cross-links. To be maximally effective, inhibitors of cell wall synthesis require actively proliferating microorganisms. shows the classification of agents affecting cell wall synthesis
Beta-Lactam Antibiotics
PENICLLINS AND ITS CONGENERS
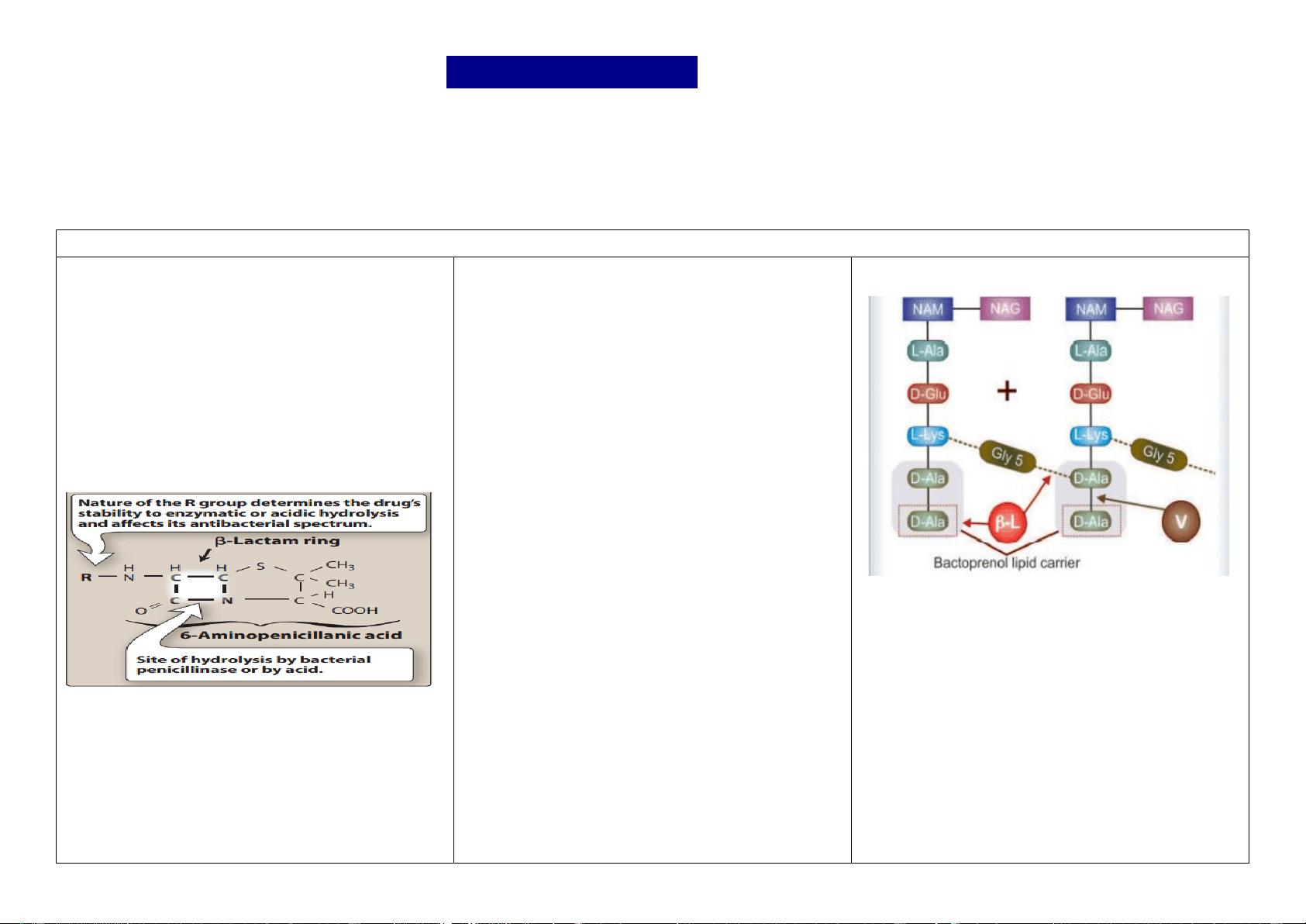
Penicillin was the first antibiotic to be used clinically in 1941. It is a miracle that the least toxic drug of its kind was the first to be discovered. Penicillin was originally obtained from the fungus Penicillium notatum, but the present source is a high yielding mutant of P. chrysogenum. Chemistry and properties the penicillin nucleus consists of fused thiazolidine and β-lactam rings to which side chains are attached through an amide linkage. Penicillin G (PnG), having a benzyl side chain at R (benzyl penicillin), is the original penicillin used clinically. The side chain of natural penicillin can be split off by an amidase to produce 6-aminopenicillanic acid. Other side chains can then be attached to it resulting in different semisynthetic penicillins with unique antibacterial activities and different pharmacokinetic profiles. At the carboxyl group attached to the thiazolidine ring, salt formation occurs with Na+ and K+. Unitage 1 U of crystalline sod. benzyl penicillin = 0.6 μg of the standard preparation. Accordingly, 1 g = 1.6 million units or 1 MU = 0.6 g. Mechanism of action Penicillins interfere with the last step of bacterial cell wall synthesis, which is the cross-linking of adjacent peptidoglycan strands by a process known as transpeptidation. Since penicillins structurally resemble the terminal portion of the peptidoglycan strand, they compete for and bind to enzymes called penicillin-binding proteins (PBPs), which catalyze transpeptidase and facilitate cross- linking of the cell wall. The result is the formation of a weakened cell wall and ultimately cell death. For this reason, penicillins are regarded as bactericidal and work in a time-dependent fashion. When susceptible bacteria divide in the presence of a β-lactam antibiotic—cell wall deficient (CWD) forms are produced. Because the interior of the bacterium is hyperosmotic, the CWD forms swell and burst → bacterial lysis occurs. This is how β- lactam antibiotics exert bactericidal action. Under certain conditions and in case of certain organisms, bizarre shaped or filamentous forms, which are incapable of multiplying, result. Grown in hyperosmotic medium, globular ‘giant’ forms or protoplasts are produced. Lytic effect of these antibiotics may also be due to derepression of some bacterial autolysins which normally function during cell division. Cross linking of peptidoglycan residues of neighbouring strands by cleavage of terminal D- alanine (D-Ala) and transpeptidation with the chain of 5 glycine (Gly5) residues. The β-lactam antibiotics (β-L) block cleavage of terminal D-Ala and transpeptidation. The peptidoglycan units are synthesized within the bacterial cell and are transported across the cell membrane by attachment to a bactoprenol lipid carrier for assembly into strands. Vancomycin (V) binds tightly to the terminal D-Ala D-Ala sequence and prevents its release from the carrier, so that further transpeptidation cannot take place
treatment with penicillin for syphilis, JHR is reported to start approximately at 4 hours, peak at 8 hours, and subside by 16 hours. Resistance Survival of bacteria in the presence of β-lactam antibiotics occurs due to the following.
1. - Lactamase production: This family of enzymes hydrolyzes the cyclic amide bond of the β-lactam ring, which results in loss of bactericidal activity. They are the major cause of resistance to the penicillins and are an increasing problem. β-Lactamases either are constitutive, mostly produced by the bacterial chromosome or, more commonly, are acquired by the transfer of plasmids. Some of the β-lactam antibiotics are poor substrates for β-lactamases and resist hydrolysis, thus retaining their activity against β-lactamase– producing organisms. [Note: Certain organisms may have chromosome-associated β-lactamases that are inducible by β-lactam antibiotics (for example, second- and third-generation cephalosporins).] Gram-positive organisms secrete β-lactamases extracellularly, whereas gram-negative bacteria inactivate β-lactam drugs in the periplasmic space. 2. Decreased permeability to the drug: Decreased penetration of the antibiotic through the outer cell membrane of the bacteria prevents the drug from reaching the target PBPs. In grampositive bacteria, the peptidoglycan layer is near the surface of the bacteria and there are few barriers for the drug to reach its target. Reduced penetration of drug into the cell is a greater concern in gram-negative organisms, which have a complex cell wall that includes aqueous channels called porins. An excellent example of a pathogen lacking high permeability porins is P. aeruginosa. The presence of an efflux pump which actively removes antibiotics from the site of action, can also reduce the amount of intracellular drug (for example, Klebsiella pneumoniae). 3. Altered PBPs: These are bacterial enzymes involved in the synthesis of the cell wall and in the maintenance of morphologic features of the bacterium. Antibiotic exposure can prevent cell wall synthesis and can lead to morphologic changes or lysis of susceptible bacteria. The number of PBPs varies with the type of organism. Modified PBPs have a lower affinity for β-lactam antibiotics, requiring clinically unattainable concentrations of the drug to effect inhibition of bacterial growth. This explains MRSA resistance to most commercially available β-lactam Uses Penicillin G is the drug of choice for infections caused by organisms susceptible to it, unless the patient is allergic to this antibiotic. However, use has declined very much due to fear of causing anaphylaxis. 1. Streptococcal infections Like pharyngitis, otitis media, scarlet fever, rheumatic fever respond to ordinary doses of PnG because Strep. pyogenes has not developed significant resistance. For subacute bacterial endocarditis (SABE) caused by Strep. viridans or faecalis high doses (10–20 MU i.v. daily) along with gentamicin given for 2 – 6 weeks is needed. 2. Pneumococcal infections PnG is infrequently used now for pneumococcal (lobar) pneumonia and meningitis, only if the strain is sensitive. 3. Meningococcal infections are still mostly responsive; meningitis and other infections may be treated with intravenous injection of high doses of PnG. 4. Gonorrhoea PnG has become unreliable for the treatment of gonorrhoea. 5. Syphilis T. pallidum has not shown any resistance and PnG is the drug of choice. Early and latent syphilis is treated either with daily i.m. injection of 1.2 MU of procaine penicillin for 10 days or with 1–3 weekly doses of 2. MU benzathine penicillin. For late syphilis, benzathine penicillin 2.4 MU weekly for 4 weeks is recommended. Cardiovascular and neurosyphilis requires sod. PnG 5 MU i.m. 6 hourlies for 10– 14 days followed by the above regimen. Leptospirosis: PnG 1.5 MU injected i.v. 6 hourly for 7 days is curative. 6. Diphtheria Antitoxin therapy is of prime importance. Procaine penicillin 1–2 MU daily for 10 days may be used to prevent carrier state. 7. Tetanus and gas gangrene Antitoxin and other measures are more important; PnG 6–12 MU/day is used to kill the causative organism and has adjuvant value. 8. Penicillin G may be used for rare infections like anthrax, actinomycosis, rat bite fever and those caused by Listeria monocytogenes, Pasteurella multocida. For trench mouth or acute necrotizing ulcerative gingivitis (ANUG) which is a mixed infection caused by spirochetes and fusobacteria, PnG (i.m.)/penicillin V (oral) or amoxicillin are generally combined with metronidazole. 9 Prophylactic uses (a) Rheumatic fever: Low concentrations of penicillin prevent colonization by streptococci that are indirectly responsible for rheumatic fever. Benzathine penicillin 1.2 MU every 4 weeks till 18 years of age or 5 years after an attack, whichever is more. ( b) Bacterial endocarditis: Dental extractions, endoscopies, catheterization, etc. cause bacteremia which in patients with valvular defects can cause endocarditis. PnG can afford protection, but amoxicillin is preferred now
SEMISYNTHETIC PENICILLINS
Semisynthetic penicillins are produced by chemically combining specific side chains (in place of benzyl side chain of PnG) or by incorporating specific precursors in the mould cultures. Thus, procaine penicillin and benzathine penicillin are salts of PnG and not semisynthetic penicillins. The aim of producing semisynthetic penicillins has been to overcome the shortcomings of PnG, which are:
- Poor oral efficacy. 2. Susceptibility to penicillinase. 3. Narrow spectrum of activity. 4. Hypersensitivity reactions (this has not been overcome in any preparation). ACID-RESISTANT ALTERNATIVE TO PENICILLIN-G Phenoxymethyl penicillin (Penicillin V) It differs from PnG only in that it is acid stable. Oral absorption is better; peak blood level is reached in 1 hour and plasma t½ is 30 – 60 min. The antibacterial spectrum of penicillin V is identical to PnG, but it is about 1/5 as active against Neisseria, other gram-negative bacteria and anaerobes. It cannot be depended upon for more serious infections and is occasionally used for streptococcal pharyngitis, sinusitis, otitis media, prophylaxis of rheumatic fever (when an oral drug has to be selected), less serious pneumococcal infections and trench mouth
PENICILLINASE-RESISTANT PENICILLINS
These congeners have side chains that protect the β-lactam ring from attack by staphylococcal penicillinase. However, this also partially protects the bacteria from the β-lactam ring: non penicillinase producing organisms are less sensitive to these drugs than to PnG. Their only indication is infections caused by penicillinase producing Staphylococci, which are not methicillin resistant as well. Utility of these penicillins has markedly declined, because ‘methicillin resistant Staph. aureus’ (MRSA) have become universally prevalent. These drugs are not resistant to β-lactamases produced by gram negative bacteria. Methicillin It is staphylococcal penicillinase resistant but not acid resistant. Because MRSA have emerged in many areas, and because methicillin caused haematuria, albuminuria and interstitial nephritis, it is no longer used. Cloxacillin/Dicloxacillin It has an isoxazolyl side chain and is highly penicillinase as well as acid resistant. Activity against PnG sensitive organisms is weaker, and it should not be used as a substitute of PnG for any other infection. activity against penicillinase producing Staph, is greater than that of methicillin, but not against MRSA. Cloxacillin is incompletely but dependably absorbed from oral route, especially if taken in empty stomach. It is > 90% plasma protein bound. Elimination occurs primarily by kidney, also partly by liver. Plasma t½ is about 1 hour. EXTENDED SPECTRUM PENICILLINS These semisynthetic penicillins are in addition active against a variety of gram-negative bacilli because of improved ability to penetrate through their cell membrane. However, they are susceptible to several b-lactamases. Semisynthetic penicillins are grouped according to their spectrum of activity
1. Aminopenicillins
This group includes ampicillin, its prodrug bacampicillin, and amoxicillin.
. Its combination with clavulanic acid extends efficacy to cover b-lactamase producing strains. For the treatment of serious Pseudomonas infections, it is often used along with gentamicin.
3. Ureidopenicillins
Piperacillin
This antipseudomonal penicillin is about 8 times more active than carbenicillin. In addition, it has good activity against Klebsiella, many Enterobacteriaceae and some Bacteroides. Piperacillin is frequently employed for treating serious gram-negative infections in neutropenic/immunocompromised or burn patients. Elimination t½ is 1 hr. It is combined with tazobactam to cover b-lactamase producing strains. (Concurrent use of gentamicin or tobramycin is advised
BETA-LACTAMASE INHIBITORS
β-lactamases are a family of enzymes produced by many gram-positive and gram-negative bacteria that inactivate β-lactam antibiotics by opening the β-lactam ring. Different β-lactamases differ in their substrate affinities. Three inhibitors of this enzyme clavulanic acid, sulbactam and tazobactam
Clavulanic acid
Obtained from Streptomyces clavuligerus, it has a β-lactam ring but no/weak antibacterial activity of its own. It inhibits a wide variety (class II to class V) of β-lactamases (but not class I cephalosporinase) produced by both gram-positive and gram-negative bacteria. Clavulanic acid is a ‘progressive’ inhibitor, because binding with β-lactamase is reversible initially, but becomes covalent later—inhibition increases with time. Called a ‘suicide’ inhibitor, it gets inactivated after binding to the enzyme. Clavulanate permeates the outer layers of the cell wall of gram- negative bacteria and inhibits the periplasmically located β-lactamase
. Pharmacokinetics Clavulanic acid has rapid oral absorption and a bioavailability of 60%. It can also be injected. The elimination t½ of 1 hr and tissue distribution matches amoxicillin, with which it is combined (called coamoxiclav). However, clavulanate is eliminated mainly by glomerular filtration and its excretion is not affected by probenecid. Moreover, it is largely hydrolysed and decarboxylated before excretion, while amoxicillin is primarily excreted unchanged by tubular secretion. Uses Addition of clavulanic acid re-establishes the activity of amoxicillin against β-lactamase producing resistant Staph. aureus (but not MRSA that have altered PBPs), H. influenzae, N. gonorrhoeae, E. coli, Proteus, Klebsiella, Salmonella and Shigella. Though Bact. fragilis and Branhamella catarrhalis are not responsive to amoxicillin alone, they are inhibited by the combination. Clavulanic acid does not potentiate the action of amoxicillin against strains that are already sensitive to it Adverse effects same as for amoxicillin alone; but g.i. tolerance is poorer—especially in children. Other adverse effects are Candida stomatitis/vaginitis and rashes. Some cases of hepatic injury have been reported with the combination
Sulbactam
It is a semisynthetic β-lactamase inhibitor, related chemically as well as in activity to clavulanic acid. It is also a progressive inhibitor, highly active against class II to V but poorly active against class I β-lactamase. On weight basis, it is 2–3 times less potent than clavulanic acid for most types of the enzyme, but the same level of inhibition can be obtained at the higher concentrations achieved clinically. Sulbactam does not induce chromosomal β-lactamases, while clavulanic acid can induce some of them. Oral absorption of sulbactam is inconsistent Therefore, it is preferably given parenterally. It has been combined with ampicillin for use against β- lactamase producing resistant strains. Absorption of its complex salt with ampicillin—sultamicillin tosylate is better, which is given orally. Indications are:
- PPNG gonorrhoea; sulbactam per se also inhibits N. gonorrhoeae.
- Mixed aerobic-anaerobic infections, intraabdominal, gynaecological, surgical and skin/ soft tissue infections, especially those acquired in the hospital.
Tazobactam
Another β-lactamase inhibitor similar to sulbactam, whose pharmacokinetics matches with piperacillin. Tazobactam is combined with piperacillin for use in severe infections like peritonitis, pelvic/urinary/ respiratory infections caused by β-lactamase producing bacilli. However, the combination is not active against piperacillin-resistant Pseudomonas, because tazobactam (like clavulanic acid and sulbactam) does not inhibit inducible chromosomal β-lactamase produced by Pseudomonas and other Enterobacteriaceae.
CEPHALOSPORINS These are a group of semisynthetic antibiotics derived from ‘cephalosporin-C’ obtained from a fungus Cephalosporium. Chemistry They are chemically related to penicillins; the nucleus consists of a β-lactam ring fused to a dihydrothiazine ring, (7- aminocephalosporanic acid). By addition of different side chains at position 7 of β-lactam ring (altering spectrum of activity) and at position 3 of dihydrothiazine ring (affecting pharmacokinetics), a large number of semisynthetic compounds have been produced Cephalosporins have been conventionally divided into 4 generations, and now some lately added members have been designated ‘5th generation All cephalosporins are bactericidal and have the same mechanism of action as penicillin, i.e., inhibition of bacterial cell wall synthesis. However, they bind to different PBPs than those which bind penicillins. This may explain differences in spectrum, potency and lack of cross resistance. Acquired resistance to cephalosporins could have the same basis as for penicillins, i.e.: a) alteration in target proteins (PBPs) reducing affinity for the antibiotic. (b) impermeability to the antibiotic or its efflux so that it does not reach its site of action. (c) elaboration of β-lactamases which destroy specific cephalosporins (cephalosporinases); the most common mechanism
FIRST GENERATION CEPHALOSPORINS
These were developed in the 1960s, have high activity against gram-positive but weaker against gram-negative bacteria.
Cefazolin It is the prototype first generation
cephalosporin that is active against most PnG sensitive organisms, i.e., Streptococci (pyogenes as well as viridans), gonococci, meningococci, C. diphtheriae, H. influenzae, clostridia and Actinomyces. Activity against Klebsiella, Moraxella catarrhalis and E. coli is relatively high, but it is quite susceptible to staphylococcal β-lactamase. Pharmacokinetics It can be given i.m. (mildly painful) as well as i.v. and has a longer t½ (2 hours) due to slower tubular secretion. Cefazolin attains higher concentration in plasma and in bile. Use It is the preferred parenteral first-generation cephalosporin, especially for surgical prophylaxis
Cephalexin It is the most commonly used orally
effective first-generation cephalosporin, similar in spectrum to cefazolin, but less active against penicillinase producing staphylococci and H. influenzae. Plasma protein binding is low; it attains high concentration in bile and is excreted unchanged in urine; t½ ~60 min..
Cefadroxil
A close congener of cephalexin; has good tissue penetration—exerts more sustained action at the site of infection, because of which it can be given 12 hourly despite a t½ of 1 hr. It is excreted unchanged in urine; the dose needs to be reduced only if creatinine clearance is < 50 ml/min. The antibacterial activity of cefadroxil and indications are similar to those of cephalexin.
SECOND GENERATION
CEPHALOSPORINS
These were developed subsequent to the first- generation compounds and are more active against gram-negative organisms with wider coverage, including some strains resistant to first generation compounds. Few members, i.e., cefoxitin are active against anaerobes as well, but none inhibits P. aeruginosa. They are weaker than the first-generation compounds against gram positive bacteria.
Cefuroxime It is resistant to gram-negative β-
lactamases, and has high activity against organisms producing these enzymes including PPNG and ampicillin-resistant H. influenzae, while retaining significant activity on grampositive cocci and certain anaerobes, but not B. fragilis. Cefuroxime can be injected i.m., and attains relatively higher CSF levels, but has been superseded by 3rd generation cephalosporins in the treatment of meningitis. It can be employed for single dose i.m. therapy of gonorrhoea due to PPNG. For gonorrhoea 1.5 g divided at 2 sites i.m. inj + probenecid 1.0 g oral single dose. Cefuroxime axetil This ester of cefuroxime is effective orally, despite incomplete oral absorption. The activity depends on in vivo hydrolysis and release of cefuroxime. Dose: 250–500 mg BD, children half dose;
Cefaclor, it retains significant activity by the oral
route and is more active than the first generation.
, it inhibits Staph. aureus. It is used mainly for respiratory, urinary, skin and soft tissue infections.
Cefixime
It is an orally active third generation cephalosporin highly active against Enterobacteriaceae, H. influenzae, Strep. pyogenes, and is resistant to many β-lactamases. However, it is not active on Staph. aureus, most pneumococci and Pseudomonas. Pharmacokinetics Cefixime is longer acting (t½ 3 hr) and has been used in a dose of 200–400 mg BD for respiratory, urinary and biliary infections. Stool changes and diarrhoea are the most prominent side effects
Cefpodoxime proxetil
It is the orally active ester prodrug of 3rd generation cephalosporin cefpodoxime. In addition to being highly active against Enterobacteriaceae and streptococci, it inhibits Staph. aureus. It is used mainly for respiratory, urinary, skin and soft tissue infections
Cefdinir
This orally active 3rd generation cephalosporin has good activity against many β-lactamase producing organisms. Most respiratory pathogens including gram-positive cocci are susceptible. Indications of cefdinir are pneumonia, acute exacerbations of chronic bronchitis, ENT and skin infections
Ceftibuten Another oral 3rd generation cepha -
losporin, active against gram-positive and few gram-negative bacteria, but not Staph. aureus. It is stable to β-lactamases, and is indicated in respiratory and ENT infections; t½ 2–3 hours.
Ceftamet pivoxil This ester prodrug of ceftamet,
a 3rd generation cephalosporin has high activity against gram-negative bacteria, especially Enterobacteriaceae and N. gonorrhoea. Ceftamet pivoxil is indicated in respiratory, skin and soft tissue infections, etc.
FOURTH GENERATION
CEPHALOSPORINS
The distinctive feature of this newer subgroup of cephalosporins is non-susceptibility to inducible chromosomal β lactamases produced by some resistant bacteria alongwith high potency against Enterobacteriaceae and spectrum of activity resembling the 3rd generation compounds.
Cefepime this 4th generation cephalosporin has
antibacterial spectrum similar to that of 3rd generation compounds, but is highly resistant to β- lactamases, hence active against many bacteria resistant to the earlier drugs. Ps. aeruginosa, Strep. pneumoniae, H. influenzae and Staph. aureus is also inhibited but not MRSA. Uses Due to high potency and extended spectrum, it is effective in many serious infections like hospital-acquired pneumonia, febrile neutropenia, bacteraemia, septicaemia. Higher concentrations are attainedin the CSF, and it is excreted by the kidney with a t½ of 2 hours.
Cefpirome This 4th generation cephalosporin is
indicated for the treatment of serious and resistant hospital-acquired infections including septicaemias, lower respiratory tract infections, etc. Its zwitterion character permits better penetration through porin channels of gram-negative bacteria. Cefpirome is resistant to many β-lactamases; inhibits type 1 β-lactamase producing Enterobacteriaceae and it is more potent against gram-positive and some gram-negative bacteria than the 3rd generation compounds.
FIFTH GENERATION CEPHALOSPORINS
The recently developed members of this new group are distinguished by their ability to kill MRSA and some other bacteria which developed penicillin resistance by producing altered PBPs. Thus, these antibiotics are effective in many resistant and hospital acquired infections. They are also designated ‘cephalosporins with antiMRSA activity’.
Ceftaroline fosamil
It is a prodrug which after i.v. infusion is rapidly converted by phosphatases to the active ceftaroline that is cidal to many gram +ive and gram
- ive bacteria including MRSA, penicillin resistant Strep. pneumoniae and Enterococcus faecalis, etc. Ceftaroline owes its activity to its ability to bind altered PBPs expressed in these bacteria. It has high affinity for PBP2a (in MRSA), for PBP2b and PBP2x (in penicillin resistant Strep. pneumoniae). It thus interferes with transpeptidation step of bacterial cell wall synthesis and exerts lethal effect. However, it is susceptible to extended-spectrum and some other b-lactamases so that organisms expressing these enzymes are nonresponsive. Uses Ceftaroline fosamil was approved for the treatment of complicated skin and soft tissue infections as well as for community acquired pneumonia (CAP), particularly those caused by MRSA, resistant Strep. pneumoniae, etc. Ceftaroline is eliminated mainly by the kidney with a t½ of 2.6 hours. Adverse effects noted are headache, dizziness, itching, rashes, fever, diarrhoea, and irritation of the injected vein.
Adverse effects Cephalosporins are generally well tolerated, but are more toxic than penicillin.
1. Pain after i.m. injection occurs with many cephalosporins, but some can be injected i.m., while others are injected only i.v. (see individual compounds). Thrombophlebitis of injected vein can occur.
- Diarrhoea due to alteration of gut ecology or irritative effect is more common with orally administered compounds like cephalexin, cefixime and parenteral cefoperazone, which is largely excreted in bile. 3. Hypersensitivity reactions are the most important adverse effects of cephalosporins. Manifestations are similar to those with penicillin, but incidence is lower. Rashes are the most frequent but anaphylaxis, angioedema, asthma and urticaria have also occurred. About 10% patients allergic to penicillin show cross reactivity with cephalosporins. Those with a history of immediate type of reactions to penicillin should better not be given a cephalosporin. Skin tests for sensitivity to cephalosporins are unreliable. A positive Coombs’ test occurs in many patients, but haemolysis is rare.
- Nephrotoxicity Some cephalosporins have low- grade nephrotoxicity which may be accentuated by preexisting renal disease, concurrent administration of an aminoglycoside or loop diuretic.
- Bleeding occurs with cephalosporins having a methylthiotetrazole or similar substitution at position 3 (cefoperazone, ceftriaxone). This is due to hypoprothrombinaemia caused by the same mechanism as warfarin and is more common in patients with cancer, intra-abdominal infection or renal failure. 6. Neutropenia and thrombocytopenia are rare adverse effects reported with ceftazidime and some others.
- A disulfiram-like interaction with alcohol has been reported with cefoperazone. Uses Currently cephalosporins are one of the most commonly used antibiotics. Among them they cover a wide range of gram-positive and gram- negative bacteria including some anaerobes, but not B. fragilis, mycobacteria and chlamydia. Recently, some anti-MRSA cephalosporins have been added to further extend their coverage. Their indications are:
- As alternatives to penicillins for ENT, upper respiratory and cutaneous infections, one of the first-generation compounds may be used.
- Respiratory, urinary and soft tissue infections caused by gram-negative organisms, especially Klebsiella, Proteus, Enterobacter, Serratia. Cephalosporins preferred for these infections are cefuroxime, cefotaxime, ceftriaxone. CAP due to penicillin-resistant Strep. pneumoniae can now be treated by ceftaroline fosamil and HAP by ceftobiprole medocaril.
- Penicillinase producing staphylococcal infections. Complicated skin and soft tissue infections due to MRSA can now be treated by ceftaroline fosamil. 4. Septicaemias caused by gram-negative organisms: an aminoglycoside may be combined with a cephalosporin. 5. Surgical prophylaxis: the first generation cephalosporins are popular drugs. Cefazolin (i.m. or i.v.) is employed for most types of surgeries including those with surgical prosthesis such as artificial heart valves, artificial joints, etc. 6. Meningitis: Optimal therapy of pyogenic meningitis requires bactericidal activity in the CSF, preferably with antibiotic concentrations several times higher than the MBC for the infecting organism. For empirical therapy before bacterial diagnosis, i.v. cefotaxime/ ceftriaxone is generally combined with ampicillin or vancomycin or both. Ceftazidime + gentamicin is the most effective therapy for Pseudomonas meningitis. All oral 3rd generation cephalosporins achieve low CSF levels, and are not useful in meningitis. 7. Gonorrhoea caused by penicillinase producing organisms: ceftriaxone is a first-choice drug for single dose therapy of gonorrhoea if the penicillinase producing status of the organism is not known, but higher dose is now recommended due to resistance. Combination with azithromycin is also advised. Cefuroxime and cefotaxime have also been used for this purpose. For chancroid also, a single dose is as effective as erythromycin given for 7 days. Ceftriaxone is an alternative drug for syphilis. 8. Typhoid: Currently, ceftriaxone and cefoperazone injected i.v. are the fastest acting and most reliable drugs for enteric fever. They are preferred over fluoroquinolones (especially in children) for empirical therapy, since many S. typhi strains are resistant to chloramphenicol, ampicillin, cotrimoxazole, and lately FQs. Advantages of ceftriaxone/cefoperazone in treating typhoid fever are: • Quick defervescence, usually in 2-3 days. • Early abetment of symptoms. • Low risk of relapse and complications. • Prevention of carrier state due to cidal action on the bacilli. • Can be used to treat typhoid carriers. 9. Mixed aerobic-anaerobic infections in cancer patients, those undergoing colorectal surgery, obstetric complications: cefuroxime, cefaclor or one of the third-generation compounds is used. 10. Hospital acquired infections, especially respiratory and other infections in intensive care units, resistant to commonly used antibiotics. Cefotaxime, ceftizoxime or a fourth-generation cephalosporin may be combined with vancomycin. Ceftobiprole medocaril is an alternative.
DAPTOMYCIN
Daptomycin is a bactericidal concentration-
dependent cyclic lipopeptide antibiotic that is an alternative to other agents, such as vancomycin or linezolid, for treating infections caused by resistant gram-positive organisms, including MRSA and vancomycin-resistant enterococci (VRE). Daptomycin is indicated for the treatment of complicated skin and skin structure infections and bacteremia caused by S. aureus, including those with right-sided infective endocarditis. Efficacy of treatment with daptomycin in left-sided endocarditis has not been demonstrated. Additionally, daptomycin is inactivated by pulmonary surfactants; thus, it should never be used in the treatment of pneumonia. Daptomycin is dosed IV once daily.
LIPOGLYCOPEPTIDES
Telavancin oritavancin and dalbavancin are
bactericidal concentration-dependent semisynthetic lipoglycopeptide antibiotics with activity against gram- positive bacteria. Spectrum The lipoglycopeptides maintain a spectrum of activity similar to vancomycin, affecting primarily staphylococci, streptococci, and enterococci. Because of structural differences, they are more potent than vancomycin and may have activity against vancomycin- resistant isolates. Mechanism of action Like vancomycin, these agents inhibit bacterial cell wall synthesis. The lipid tail is essential in anchoring the drug to the cell walls to improve target site binding. Additionally, telavancin and oritavancin disrupt membrane potential. In combination, these actions improve activity and minimize selection of resistance.in treating acute bacterial skin and skin structure infections (ABSSSI) and hospital-acquired pneumonia caused by resistant gram-positive organisms, including MRSA. Adverse effect profile which includes nephrotoxicity, risk of fetal harm, and interactions with medications known to prolong the QTc interval (for example, fluoroquinolones and macrolides). Prior to initiation, assessment of renal function, pregnancy status, and current medications is needed to ensure safe administration. In contrast to telavancin, oritavancin, and dalbavancin have prolonged half-lives (245 and 187 hours, respectively), allowing for single-dose administration for the management of ABSSSI.