Download Antivirals ( RetroVirals ) and more Study notes Pharmacology in PDF only on Docsity!
Antiviral Drugs (Anti-retrovirus)
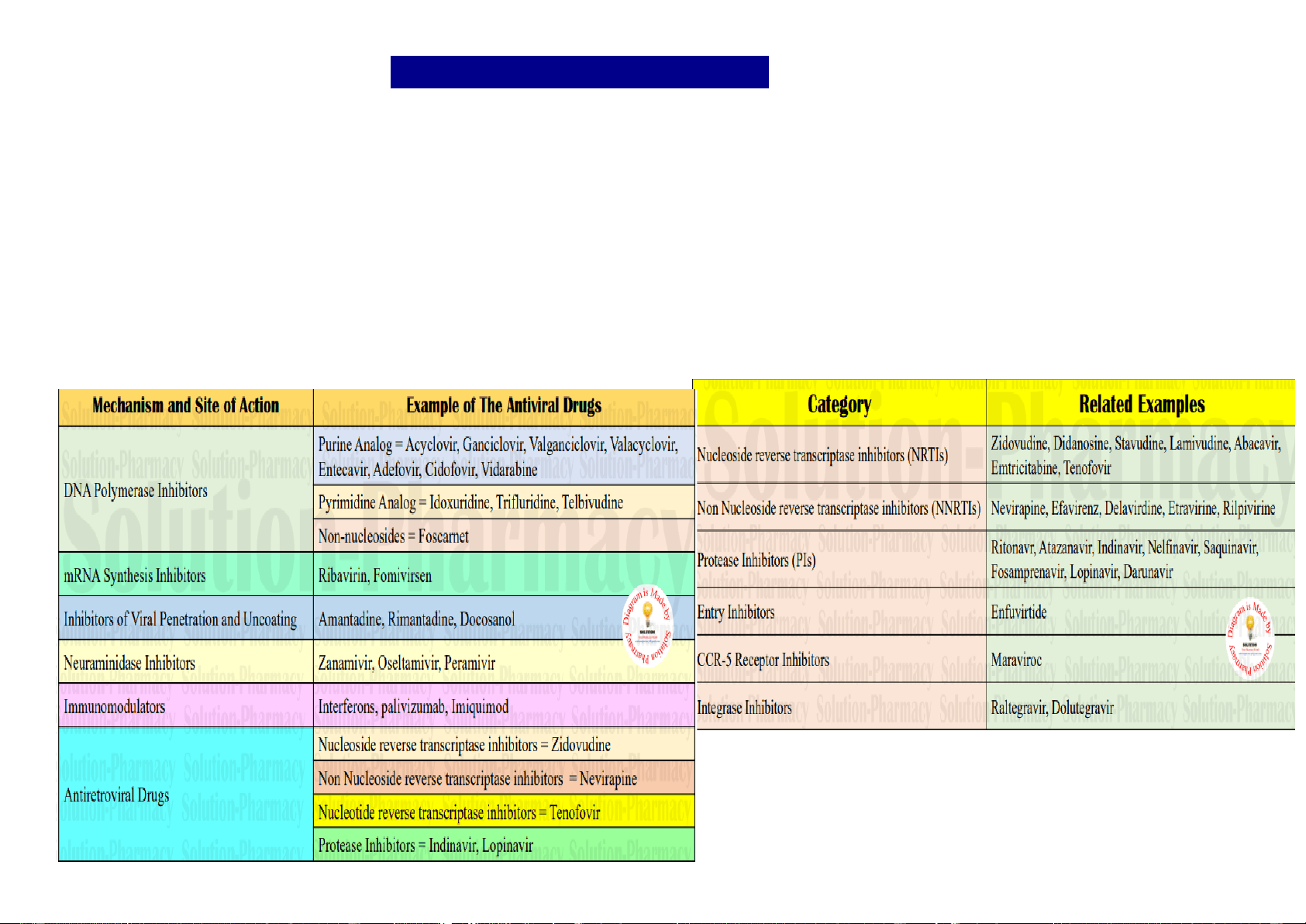
A retrovirus is a type of virus that inserts a DNA copy of its RNA genome into the DNA of a host cell that it invades, thus changing the genome of that cell. HIV is a single stranded RNA retrovirus which uniquely carries out reverse transcription of proviral DNA from viral RNA (normally RNA is transcripted from DNA) with the help of a viral RNA-dependent DNA polymerase (reverse transcriptase). The primary cell type attacked by HIV is the CD4+ helper T-lymphocyte, but later macrophages and some other cell types may also be infected. When population of CD4 cells declines markedly These are drugs active against human immunodeficiency virus (HIV) which is a retrovirus. They are useful in prolonging and improving the quality of life and postponing complications of acquired immunodeficiency syndrome (AIDS) or AIDS-related complex (ARC), but do not cure the infection. The clinical efficacy of antiretrovirus drugs is monitored primarily by plasma HIV-RNA assays and CD4 lymphocyte count carried out at regular intervals. The two established targets for anti-HIV attack are: (a) HIV reverse transcriptase: Which transcripts HIV-RNA into proviral DNA. (b) HIV protease: Which cleaves the large virus directed polyprotein into functional viral proteins.
- Fusion of viral envelope with plasma membrane of CD4 cells through which HIV-RNA enters the cell
- Chemokine coreceptor (CCR5) on host cells which provide anchorage for the surface proteins of the virus.
- HIV-integrase: Viral enzyme which integrates the proviral DNA into host DNA.
Life cycle of HIV. Binding of viral glycoproteins to host cell CD4 and chemokine receptors leads to fusion of the viral and host cell membranes via gp41 and entry of the virion into the cell. After uncoating, reverse transcription copies the single-stranded HIV RNA genome into double-stranded DNA, which is integrated into the host cell genome. Gene transcription by host cell enzymes produces messenger RNA, which is translated into proteins that assemble into immature noninfectious virions that bud from the host cell membrane. Maturation into fully infectious virions is through proteolytic cleavage. NNRTIs, nonnucleoside reverse transcriptase inhibitors; NRTIs, nucleoside/nucleotide reverse transcriptase inhibitors
Side effects are few—headache, fatigue, rashes, nausea, anorexia, abdominal pain. Pancreatitis and neuropathy are rare. Hematological toxicity does not occur.
Abacavir
Mechanism of action This guanosine analogue is a clinically potent ARV drug that acts after intracellular conversion to cabovIr triphosphate, which gets incorporated in proviral DNA and terminates chain elongation. Rapid reduction in plasma HIV-RNA count and rapid rise in CD4 cell count has been noted when abacavir was given to AIDS patients. Resistance to ABC develops slowly, and it exhibits little cross resistance with other NRTIs. Pharmacokinetics Its oral bioavailability is 80% and it is mainly eliminated by metabolism. The plasma t½ is 1–1. hour, but intracellular t½ of the active metabolite is
12 hours Hypersensitivity reactions such as rashes, fever, abdominal pain, bowel upset, flu-like respiratory and constitutional symptoms occur in 2 – 5% adult patients and are the major problems. A genetic basis and massive release of TNFα have been related to this reaction. Abacavir should never be given again to a patient who has developed this reaction. It should also be avoided in patients with cardiovascular risk factors. side effects Lypodystrophy is least likely. Avoidance of alcohol is advised. Combination regimens including abacavir are frequently used, and it is a component of the preferred 1st line WHO regimen for children.
Tenofovir (TDF)
This is the only nucleotide (not nucleoside) analogue that is a commonly used anti-HIV drug. It is also active against HBV.Tenofovir was initially used only in previously treated patients, but because of good tolerability profile, it is now being included in first line regimens as well. Tenofovir containing regimens have been found at least as effective and less toxic as other first line regimens. It is included in the preferred 1st line WHO regimen for adults and adolescents. Renal toxicity is to be watched.
Emtricitabine (FTC)
Mechanism of action It is a fluorinated cytidine analogue which is converted intracellularly by cellular kinases into its triphosphate which acts as the HIV reverse transcriptase inhibitor. Pharmacokinetics Emtricitabine is well absorbed orally, little metabolized and largely excreted unchanged by the kidney with a t½ of 10 hrs. However, the intracellular t½ of the active triphosphate is much longer (~40 hrs), permitting once daily dosing. Dose reduction is needed in renal impairment. Resistance Some cross resistance with lamivudine occurs, but not with other NRTIs. Uses Like lamivudine, it is also active against HBV, but should not be used in HIV-HBV coninfected patients because sudden stoppage of therapy can cause rebound exacerbation of hepatitis. Emtricitabine is one of the 1st line anti-HIV drugs, and is a component of the preferred WHO regimen for treating adults and adolescent HIV patients. In combination with tenofovir, it is advocated by WHO for pre-exposure prophylaxis of HIV in high-risk adults. Emtricitabine is one of the least toxic ARV drugs; side effects fatigue, headache, nausea, diarrhoea, and discoloration of exposed skin. It is marketed only as fixed dose combination tablets
Non-nucleoside reverse transcriptase
inhibitors (NNRTIs)
Nevirapine (NVP), Efavirenz (EFV)
Mechanism of action These are nucleoside unrelated compounds which directly inhibit HIV reverse transcriptase without the need for intracellular phosphorylation. Their locus of action on the enzyme is different from that of NRTIs, and they are non-competitive inhibitors. They are more potent than AZT on HIV-1, but do not inhibit HIV-2. Accordingly, they are not indicated in infections caused by HIV-2 (HIV- 2 infections are infrequent and milder). If used alone, viral resistance to NNRTIs develops rapidly by point mutation of the enzyme; they should always be combined between NVP and EFV is common, but not with NRTIs or PIs. A patient failing a regimen containing NVP should not be treated with EFV and vice versa, but can be put on etravirine regimen. Pharmacokinetics NVP is well absorbed orally; is extensively metabolized, mainly by CYP3A4 and to a lesser extent by CYP2B6, with a t½ of ~ 30 hours. Oral absorption of EFV is ~ 50%, but the t½ is longer ( hours). It is completely metabolized, mainly by CYP2B6 and a smaller fraction by CYP3A4. Both are enzyme inducers, and cause autoinduction of their own metabolism. However, EFV inhibits CYP3A4 as well Rifampin induces NVP metabolism and makes it ineffective, but has little effect on EFV levels. If a patient being treated with NVP develops TB and is put on rifampin, Uses NVP should be replaced by EFV. The NNRTIs are indicated in combination regimens for HIV. Either NVP or EFV is included in the first line triple drug regimen used by NACO, but the WHO (2016) regimen prefers EFV and advocates NPV as an alternative. These drugs have succeeded in reducing HIV-RNA levels when an earlier regimen (not including an NNRTI) has failed.
Rashes are the commonest adverse effect, followed by nausea and headache. Occasionally skin reactions are severe. Fever and rise in transaminases occur dose dependently. NVP is potentially hepatotoxic. In patients developing NVP toxicity, it should be replaced by EFV which has low hepatotoxicity. NVP should not be used in patients with hepatic dysfunction. Efavirenz (EFV) Its side effects are headache, rashes, dizziness, insomnia and a variety of neuropsychiatric symptoms. However, these symptoms decrease over time and discontinuation rate (due to adverse effect) is low. Gynaecomastia can occur. On the basis of animal studies, EFV was considered to be teratogenic, but recent human data have exonerated it; and the WHO (2016) regimen includes EFV in the preferred 1st line regimen for pregnant and breast-feeding women. Because of its longer plasma t½, occasional missed doses of EFV are less damaging.
Etravirine
This is a second generation NNRTI which is active against HIV-1 mutants that are resistant to other NNRTIs. Etravirine is indicated in adults and children >6 yr who have already been treated and are resistant/intolerant to other NNRTIs Pharmacokinetics The oral absorption of etravirine is enhanced by food and it is completely metabolized by CYP3A4, CYP2C9 and CYP2C19 with a plasma t½ of 40 hours. Metabolites are excreted in urine. Etravirine induces CYP3A4, but inhibits CYP2C9 and 2C19. As such it interacts with many drugs. Use Etravirine is approved for use only in combination with a NRTI + one of PIs, e.g., darunavir/r or lopinavir/r or saquinavir/r, but not with fosamprenavir/r or atazanavir/r. Adverse effects are nausea and skin rashes, which can rarely be very serious
Retroviral protease inhibitors (PIs)
An aspartic protease enzyme encoded by HIV is involved in the production of structural proteins and enzymes (including reverse transcriptase and integrase) of the virus from the large viral polyprotein synthesized in the infected cell. The polyprotein is broken into various functional components by this protease enzyme. It acts at a late step in HIV replication, i.e., maturation of the new virus particles when the RNA genome acquires the core proteins and enzymes. Mechanism of action They bind to the active site of the protease molecule, interfere with its cleaving function, and are more effective viral inhibitors than AZT. The PIs do not need intracellular activation by phosphorylation. Because they act at a late step of viral cycle, they are effective in both newly as well as chronically infected cells. Under their influence, HIV-infected cells produce immature noninfectious viral progeny—hence prevent further rounds of infection Pharmacokinetics Oral bioavailability of PIs is variable (IDV and RTV ~65%, NFV >20%, SQV 15%) and their plasma t½ ranges from 2 – 8 hours. All are extensively metabolized mainly by CYP3A4, except NFV which is mainly a substrate of CYP2C19. All PIs (especially ritonavir and lopinavir) are potent inhibitors of CYP3A4, while some other CYP isoenzymes are induced. The PIs interact with many drugs. Nelfinavir, lopinavir and ritonavir induce their own metabolism Uses The PIs are not used as monotherapy, because resistance develops over months due to selection of resistant mutants. Combination of NRTIs with PIs is more effective than either drug given alone, and triple therapy is more effective than double therapy. Current recommendation is to use a PI in combination with either two NRTIs or one NRTI + one NNRTI. However, PIs are avoided in 1st line regimens, because their use in initial regimens markedly restricts second line regimen options. Most guidelines, including that of WHO and NACO, reserve them for failure cases Adverse effect the most prominent adverse effects of PIs are gastrointestinal intolerance, asthenia, headache, dizziness, limb and facial tingling, numbness and rashes. Of particular concern are lipodystrophy (abdominal obesity, buffalo hump with wasting of limbs and face), dyslipidaemia (raised triglycerides and cholesterol) which may necessitate restriction of lipids in diet. Insulin resistance occurs and diabetes may be exacerbated. Indinavir crystalises in urine and increases risk of urinary calculi Atazanavir (ATV) This PI is administered with light meal which improves absorption, while acid suppressant drugs decrease its absorption. ATV is metabolized primarily by CYP3A4, which is also moderately inhibited by it. Bioavailability and efficacy of ATV is improved by combining with RTV. The t½ is 6– 8 hours. Loose motions, nausea and abdominal pain are the side effects. Dyslipidaemia and other metabolic complications are minimal with ATV, but jaundice occurs in some patients without liver damage due to inhibition of hepatic glucuronyl transferase
Indinavir (IDV)
It is to be taken on empty stomach; g.i. intolerance is common; excess fluids must be consumed to avoid nephrolithiasis. Hyperbilirubinaemia occurs. It is infrequently used now. Nelfinavir (NFV) It is to be taken with meals, since food increases absorption, but bioavailability is erratic. NFV is mainly metabolized by CYP2C19. Often produces diarrhoea and flatulence;
Entry (fusion) inhibitor
Enfuvirtide
This HIV-derived synthetic peptide acts by binding to HIV- 1 envelope transmembrane glycoprotein (gp41) which is involved in fusion of viral and cellular membranes. Fusion of the two membranes is thus prevented and entry of the virus into the cell is blocked. It is not active against HIV-2. No cross resistance with other classes of ARV drugs occurs. Administered s.c. twice daily, it is used as add-on drug to an optimized regimen in selected patients who have failed many earlier regimens or have multidrug resistant HIV, and for whom there is no other treatment option. The injections are painful and cause local nodules/cysts. The cost and inconvenience are prohibitive.
CCR5 receptor inhibitor
Maraviroc The globular glycoprotein gp120 of
the HIV envelope anchors to the CD4 site of host cell by binding to a cell membrane receptor, which mostly is the CCR5 chemokine receptor (most HIV are CCR5-tropic). Maraviroc is a novel anti-HIV drug which targets the host cell CCR5 receptor and blocks it. Attachment of the virus and subsequent entry of viral genome into the cell is thus interfered. It has no effect on HIV strains that are CXCR4 receptor tropic), or dual CCR5/CXCR4 tropic. Added to optimized background therapy in patients who have already been treated with several regimens and who have CCR5-tropic HIV infection, maraviroc has resulted in marked reduction in HIV- RNA load, and improvement in CD4 count. It is active orally and there is no cross resistance with any other ARV drug. However, CCR5-tropism must be proven before using it. Side effects are cough, fever, abdominal pain, rashes and postural hypotension, but tolerability of maraviror in general is satisfactory PHARMACOKINETIC ENHANCERS Cobicistat Cobicistat is a pharmacokinetic enhancer or booster drug used in combination treatments for HIV. This agent inhibits CYP3A isoenzymes and is used to enhance the bioavailability of the protease inhibitors atazanavir and darunavir, and the integrase inhibitor elvitegravir Ritonavir Ritonavir is no longer used as a single PI but, instead, is used as a pharmacokinetic enhancer or “booster” of other PIs. Ritonavir is a potent inhibitor of CYP3A, and concomitant ritonavir administration at low doses increases the bioavailability of the second PI, often allowing for longer dosing intervals. The resulting higher Cmin levels of the “boosted” PI also help to prevent the development of HIV resistance. Therefore, “boosted” PIs are recommended for use in initial HIV regimens in certain clinical situations. Metabolism by CYP3A4 and CYP2D6 and biliary excretion are the primary methods of elimination. Ritonavir has a half-life of 3 to 5 hours. Although ritonavir is primarily an inhibitor of CYP isoenzymes, it may also induce several CYP isoenzymes, and numerous drug interactions have been identified. Selection of the appropriate combination is based on 1) avoidance of the use of two agents of the same nucleoside analog; 2) avoidance of overlapping toxicities and genotypic and phenotypic characteristics of the virus; 3) patient factors, such as disease symptoms and concurrent illnesses; 4) impact of drug interactions; and 5) ease of adherence to the regimen