Download Crystallography Lecture 11: Determining Initial Phases in Protein Crystallography and more Study notes Biochemistry in PDF only on Docsity!
Lecture 11^
Phase problem:Determining an initial phase angle
αfor each recorded reflectionhkl^
Methods:Heavy atom methods (isomorphous replacement Hg, Pt)Halides soaks (Br, I salts)Anomalous scattering (SAD, MAD)Molecular replacementDirect MethodsComputational maximum entropy methods
ρ(x,y,z) =
F^ cos 2hkl^
π^ (hx+ky+ lz -
α)hkl
V^ h^
k^ l
Lecture 11^
Molecular replacement:Requirement: a suitable phasing model(approximates your molecule to get a starting set of initial phases)How to get a model:Multiple sequence alignments (greater than 20% identity)Threading techniques (~70% of all protein folds known)Protein data base (+20,000 entries)Other biophysical techniques (NMR, solution scattering)
Lecture 11^
- Translation function: Determine the position of the crystalstructure to be determined (T
, T, Tx y ) relative to a model.z
The search (phasing) model is orientated to be the same as that ofthe unknown molecule in the crystal unit cell. It is then translated inreal space within the unit cell and structure factors F
andhcal αarehcal^
calculated and compared with observed values of F
.hob
(grid search -imposing crystallographic symmetry)Concept:Minimize the difference between F
and Fobs
(ie maximize thecals
agreement between the search model and observed data and calculateinitial phases. R-factor search
1,3^ X
T^
X’
2,3^ Y
+ T^
=^
Y’
3,^
Z^ T
Z’
Lecture 11^
Determining an initial phase angle
αfor each recorded reflectionhkl^
ρ(x,y,z) =
Fhkl^
cos 2π^ (hx+ky+ lz -
α)hkl
V^ h^
k^ l
Fhob^
αhcal
F^ hcal
R=work^ Σ^ Fhob
=^
measured structure factor of reflection h Fhcal^
=^
calculated structure factor of reflection h K^
=^
scaling factor
Lecture 11^
Concept:Collect native and then heavy atom soaked diffraction data set.The heavy atom ion will bind to one or a number of specific siteson the protein, without effecting the the protein conformation orcrystal packing (isomorphous). The difference between the twodata sets is only from the heavy atoms.Examples of binding sites:Cysteines
Hg
Cysteines, histidines, methionines
Pt
Specific binding must be foundTips try different ionic compounds at various pHs
Lecture 11^
Once R
is minimizedwork (~40% or lower)Dependent on resolution, model accuracy etc.The calculated
αcan be directly used as the initial phaseshcal^ for the observed F
and an electron density map can be generatedhobs
Calculation of the initial electron density map
Lecture 11^
10
Least-squares model refinement:The model/improved electron density map can be used to calculateimproved structure factorsGoal to find a set of atom positions/parameters that give F
thathcal
minimize the difference to the observed Fobsminimize
Φ^ =^ Σ^
w(|F| - |Fhob
(^2) |)hcal h Fhobs^
=^
measured structure factor Fhcal^
=^
calculated structure factor w^ =
weighting (reliability of F
)^ (1/hob
(^2) σ)h
Lecture 11^
11
Other considerations:Model Geometry:
bond lengths
bond angles
Φ^ =^ Σ^
w(|F| - |Fhob
(^2) |)+hcal Σ^ w(lideal
- l) model 2 +^ Σ^ w(a
- a^ ideal^ model
h^
l^
a torsion angles+ Σ w(t^ ideal
Always check rms deviation of model geometryAdditional refinement parameters:F
= Ghcal ∑^ n^ fejj^
-2πi(hxj+kyj+lzj)
-Bj(sin. e
(^2) θ/λ)
j n^ =occupancy, Bj^
= temperature factor of atom jj G overall scale factor of |F
| to |Fhcal
|hob
Lecture 11^
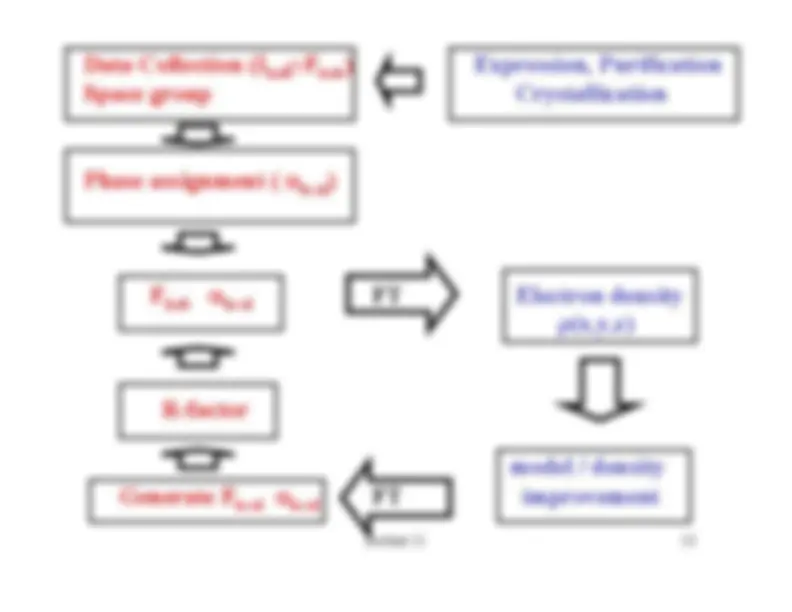
Data Collection (I
Fhob hob
)^
Expression, Purification
Space group
Crystallization
Phase assignment (
α)hcal Fαhob^
hcal^
FT^
Electron density^ ρ(x,y,z)
R-factor
model / density
Generate F
αhcal hcal
FT
improvement
Lecture 11^
Resolution (
6.^
4.^
3.^
Structure ONLY as good as the resolution of map!