


































Study with the several resources on Docsity

Earn points by helping other students or get them with a premium plan

Prepare for your exams
Study with the several resources on Docsity
Earn points to download
Earn points by helping other students or get them with a premium plan
Notes on interpreting MS/MS proteomics results
Typology: Lecture notes
1 / 72

This page cannot be seen from the preview
Don't miss anything!
technologies for separating proteins and peptides
technologies for assessing protein-protein interactions
technologies for identifying proteins*
technologies for quantifying protein expression*
bioinformatic tools for communication
Isolation of protein(s) of interest
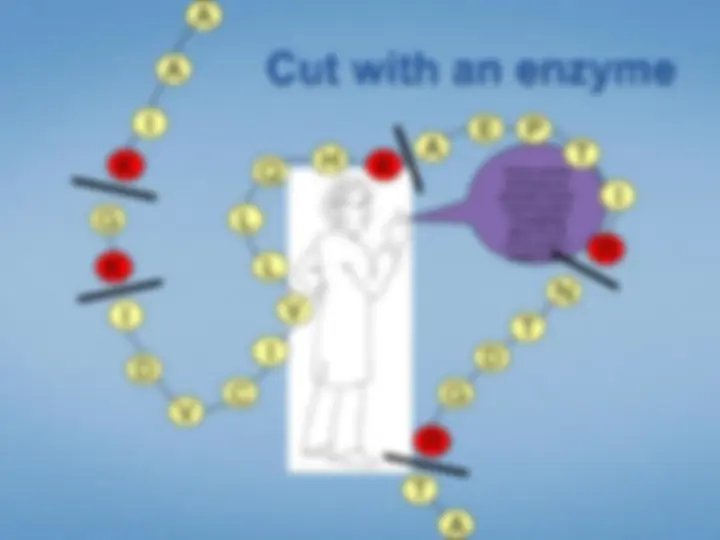
Cleavage at specific residues
MALDI-TOF analysis of resultant peptides
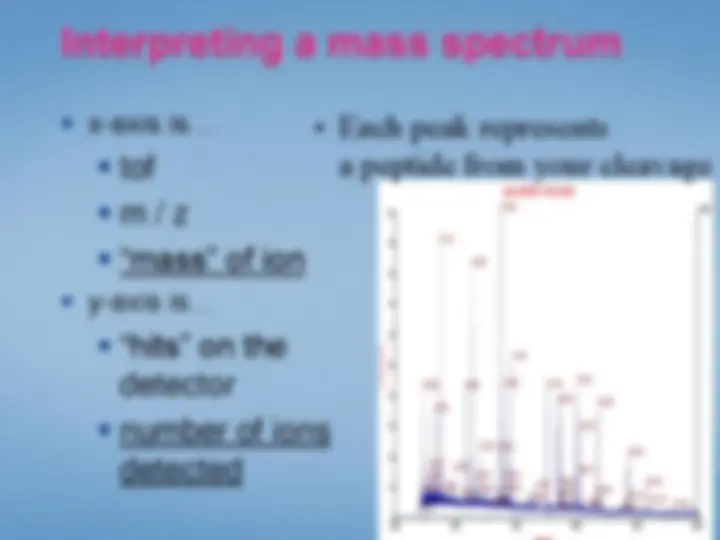
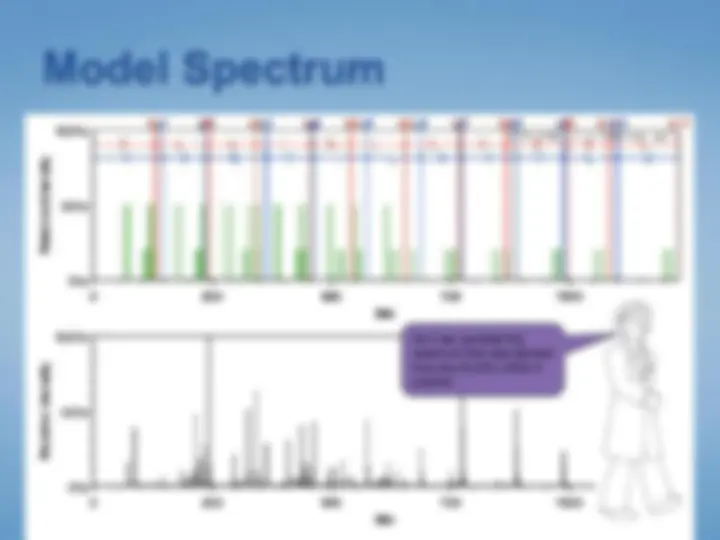
Extract peptide masses from the spectrum
Search databases for matching “peptide mass fingerprint”
Proteases: trypsin, chymotrypsin, et al.
trypsin: positive charged AA at C-terminus of peptide MALDI ionization shows bias for charged peptides
Chemical: CNBr, et al.
Other cleavage goals experimental proteases domain mapping conformational mapping
Cleveland mapping: after cleavage with a specific protease, peptide products are compared in adjacent lanes on an SDS-PAGE gel.
365.0 760.6 1156.2 (^) Mass (m /z) 1551.8 1947.4 2343.0^0
4.5E +
0
10
20
30
40
50
60
70
80
90
100
% Intensity
1053.6285^ Spec #1 MC [ B P = 1053.6, 44590]
549.3439 (^) 822.4886 1072.6033^ 1355.7562^ 1550. 617.4088^ 1426.8657^ 1690.
916.5234 (^) 1036.5975 (^) 1890. 585.3485^ 591.3327 741.4249^ 893.4636 1075.6256 1569. 537.3699 679.4092800.4719 919.5099 1088.6108 1278.6373 1436.78511443.8027 1674.6634 1893.11102008. 527.2609 1003.8684^ 1252.6669^ 1432.1174^ 1625.0525^ 1856.6127 2033.1769 2185.
Digest experimental protein with protease
Size resultant peptides
Generate list of “peak masses” (a.k.a., peptide mass fingerprint)
Archive of protein sequences “Index” via in silico digestion with protease Calculate theoretical masses of individual “peptides.” Generate a “look up table” for use by a scoring algorithm to allow ranking of “matches.”
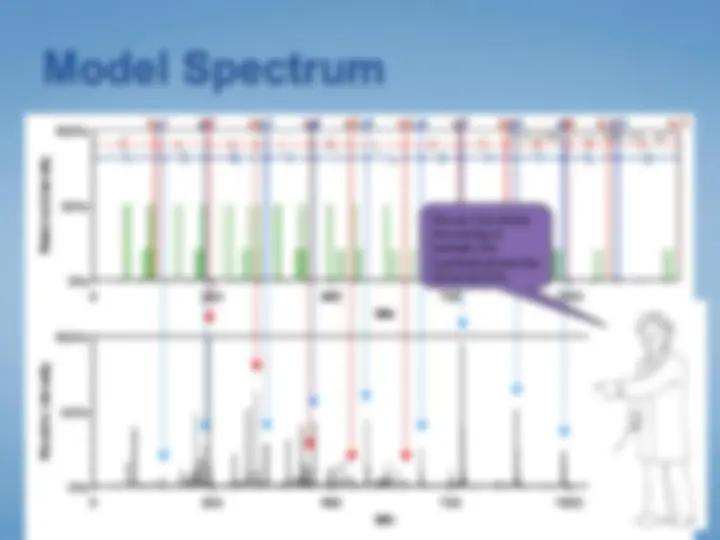
Search = “match my peptide masses”
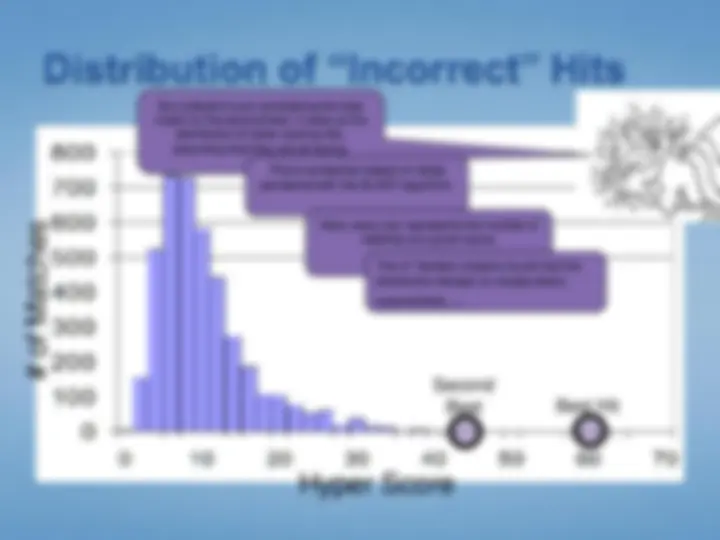
When searching, coincidental, erroneous matches will occur.
Thus, database searches generate lists of candidates, most of which are coincidental matches.
Your mission: to select the best candidate, and to justify a claim that this candidate is “correct.”
When a protein (or population of proteins) is digested, the distribution of sizes of proteolytic peptide products is not random. 0
5
10
15
20
25
30
35
40
45
900 910 920 930 940 950
peptide size
number of peptides
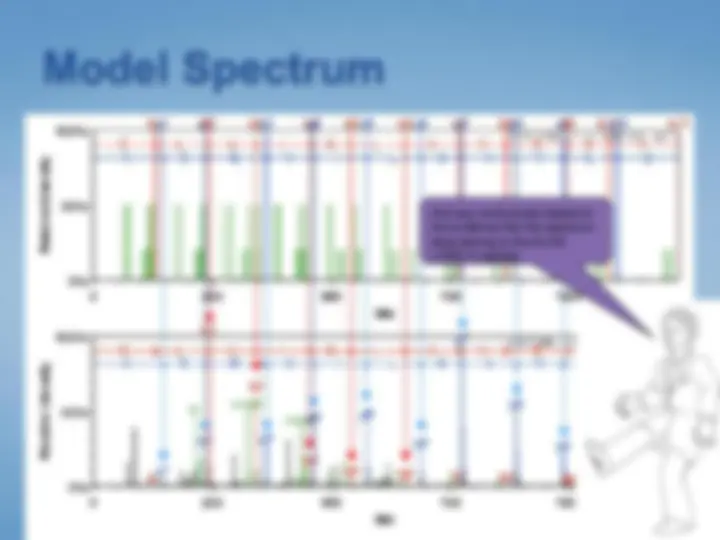
Thus, the odds of a “match” resulting from simple coincidence are lower if the peptide size is rare, and higher if the peptide size is common.
Some “matches” deserve extra impact in your scoring scheme.
0
5
10
15
20
25
30
35
40
45
900 910 920 930 940 950
number of peptides
peptide size
This is an foremost an introduction so we’re first going to talk about motivations behind the development of^ Then we’re going to talk about the the first really useful bioinformaticstechnique in our field, SEQUEST.
extended by two other tools^ This technique has been called X! Tandem andMascot.
We’re also going to talk about how these programs differ
and how we can use that to ouradvantage by considering them simultaneously using probabilities.
identifying proteins^ how you go about spectrometry in the^ with tandem mass first place
And to use this technique you generally have tolyse the protein about 8 to 20into peptides amino acids in length and…
A A I K G K I D V
C
I
V
L
L
Q H^ K^
A
E P T I R N T D G R T A
A A I K G K I D V
C
I
V
L
L
Q H^ K^
A
E P T I R N T D G R T A
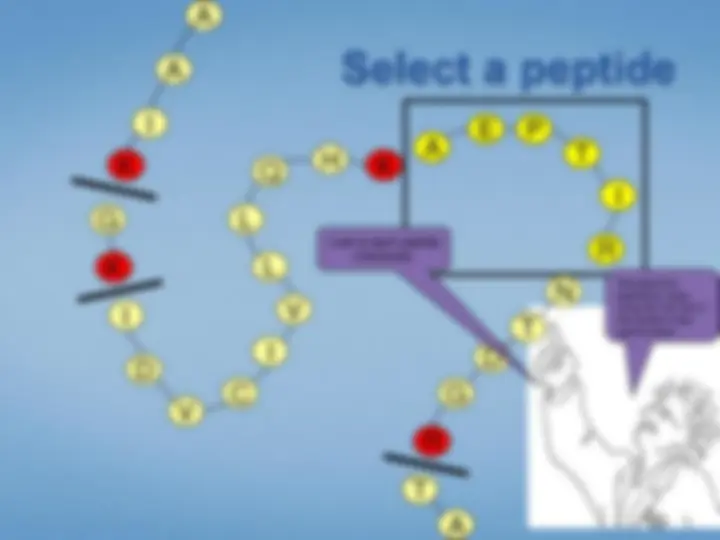
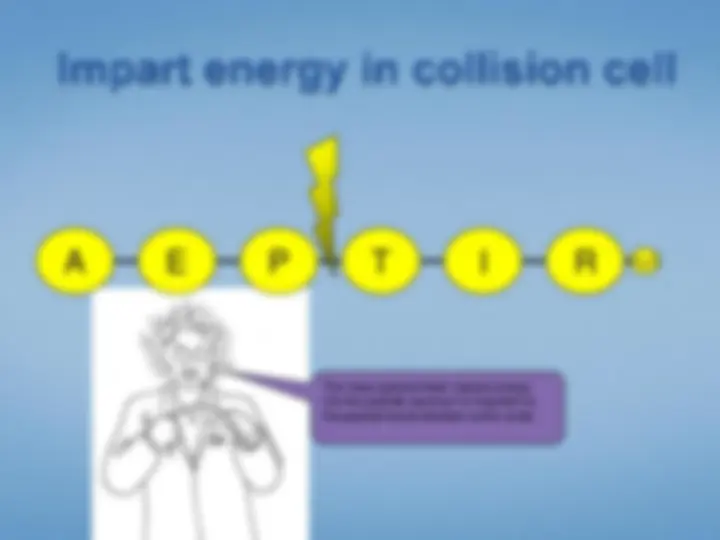
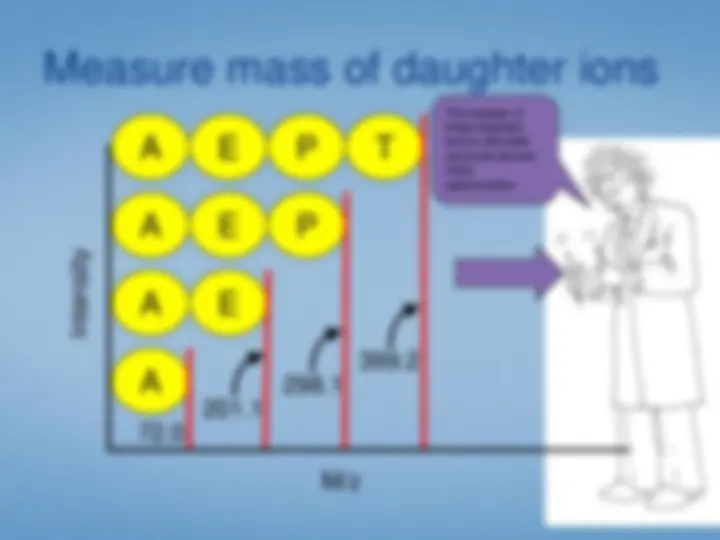
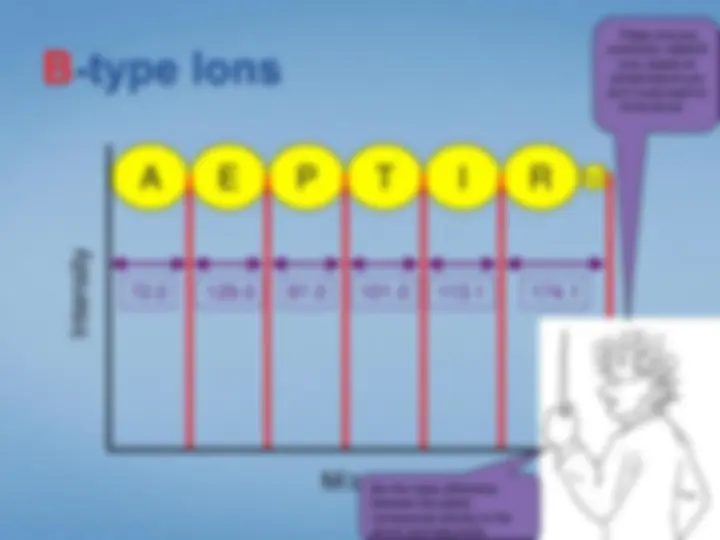
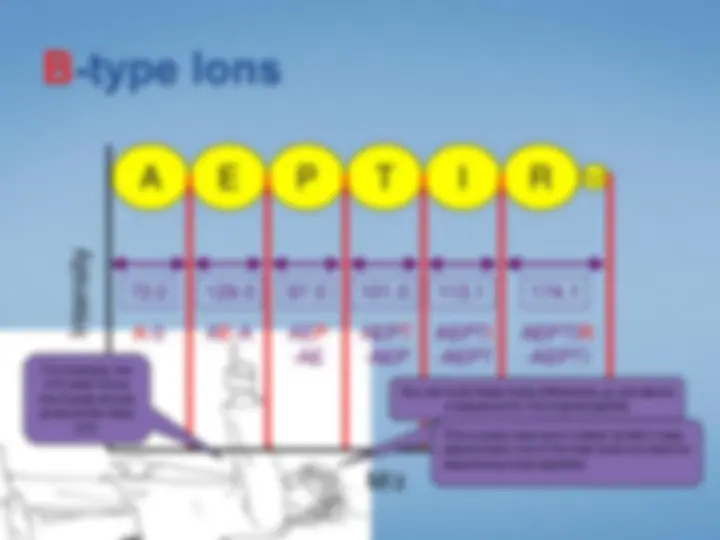
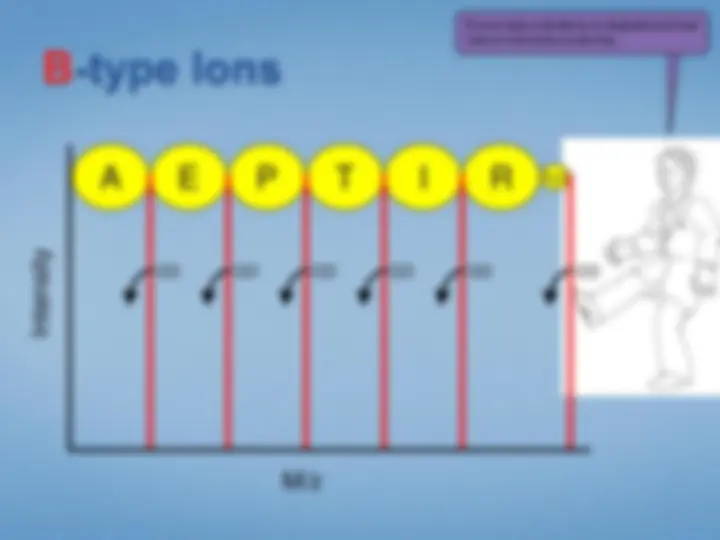
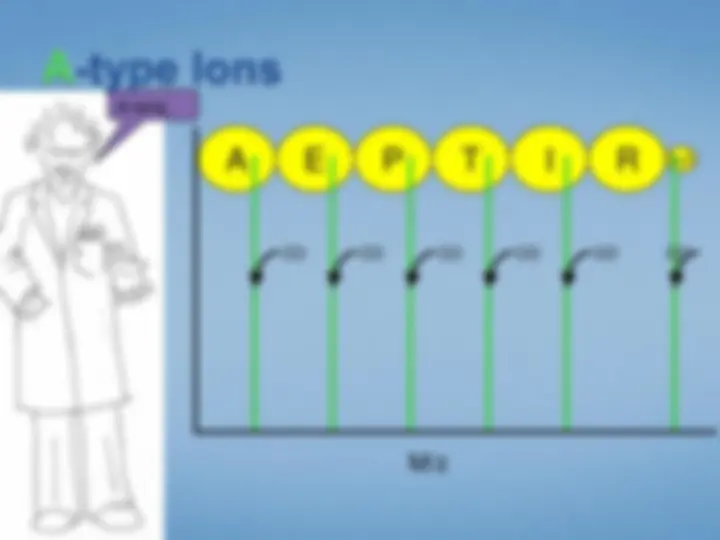
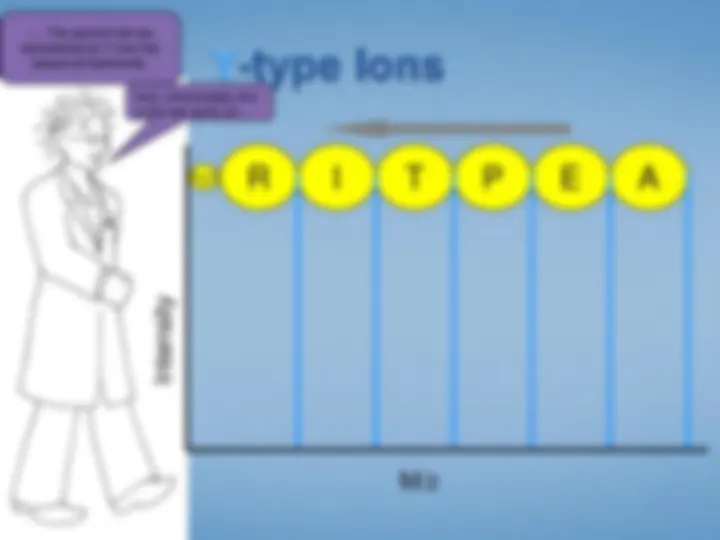
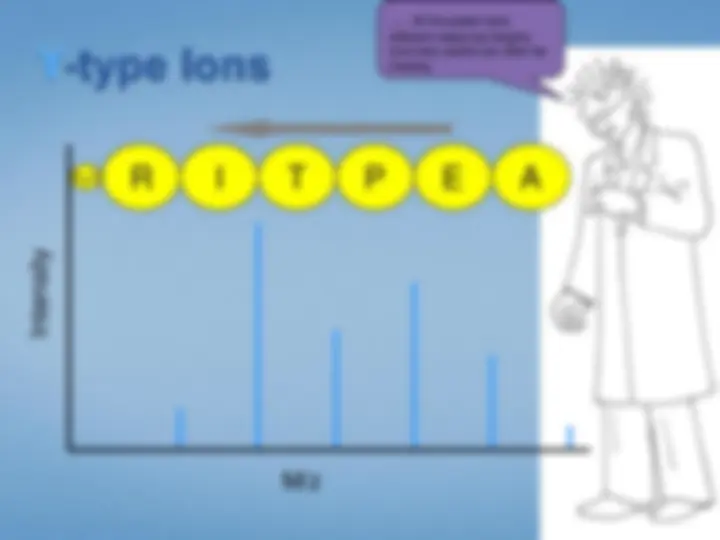
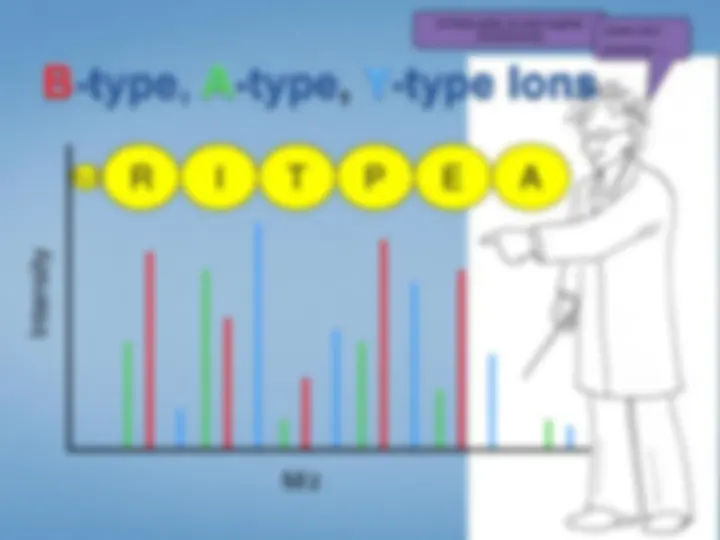
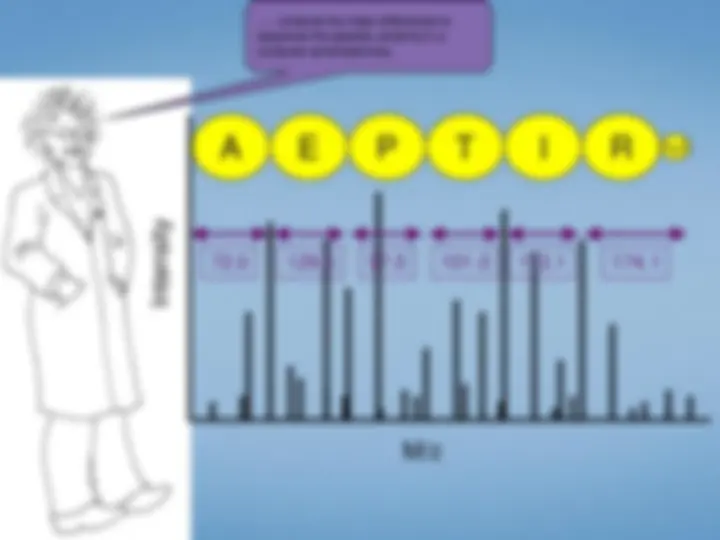
Look at each peptideindividually. We select the peptide by mass using the first half ofthe tandem mass spectrometer