



Práctica 12
Bioinformática







Prepara tus exámenes y mejora tus resultados gracias a la gran cantidad de recursos disponibles en Docsity

Gana puntos ayudando a otros estudiantes o consíguelos activando un Plan Premium

Prepara tus exámenes
Prepara tus exámenes y mejora tus resultados gracias a la gran cantidad de recursos disponibles en Docsity
Prepara tus exámenes con los documentos que comparten otros estudiantes como tú en Docsity
Encuentra los documentos específicos para los exámenes de tu universidad
Estudia con lecciones y exámenes resueltos basados en los programas académicos de las mejores universidades
Responde a preguntas de exámenes reales y pon a prueba tu preparación
Consigue puntos base para descargar
Gana puntos ayudando a otros estudiantes o consíguelos activando un Plan Premium
Comunidad
Pide ayuda a la comunidad y resuelve tus dudas de estudio
Ebooks gratuitos
Descarga nuestras guías gratuitas sobre técnicas de estudio, métodos para controlar la ansiedad y consejos para la tesis preparadas por los tutores de Docsity
Practica de Laboratorio de Bioinformatica
Tipo: Ejercicios
1 / 14

Esta página no es visible en la vista previa
¡No te pierdas las partes importantes!
Declara dos secuencias tipo cadena de caracteres que representen dos secuencias de nucleótidos : 'AGCACACA','ACACACTA'
Declara una matriz bidimensional llamada High ó H de 0s con la longitud + 1 de una de las secuencias (usa la función de la biblioteca numpy)
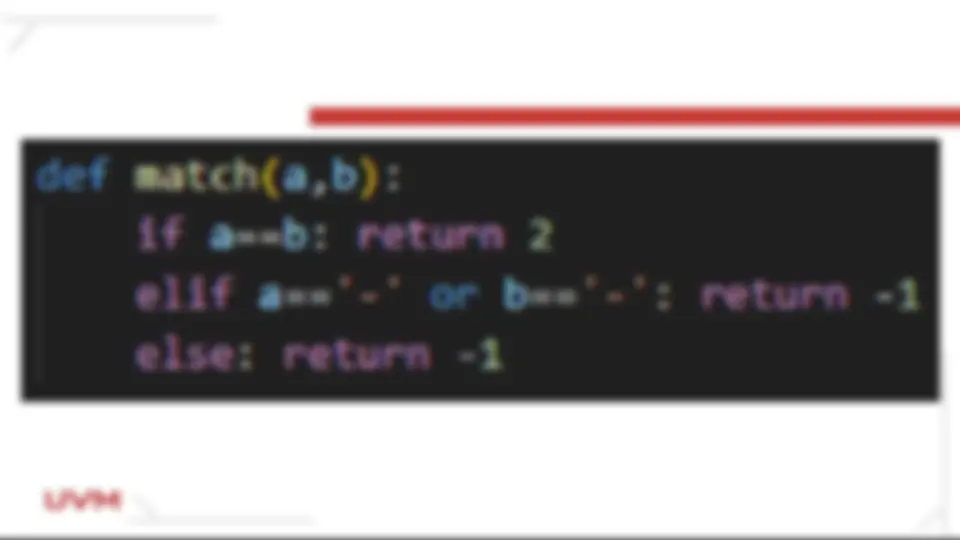
Si la posición a ó alpha es igual a la posición b ó beta, el valor que devuelde es 2 porque son iguales Si cualquiera de las posiciones es igual a '-' el valor que devuelve es - 1 (corresponde a un gap) Si las posiciones alpha o beta son diferentes la función devuelve un valor de - 1
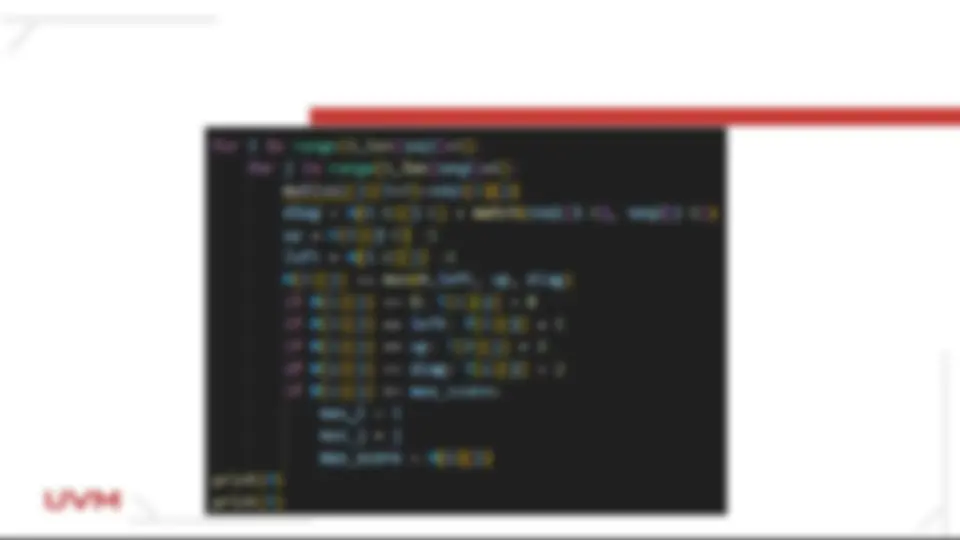
Dentro de un ciclo anidado que inicie con los renglones de la matriz bidimensional H o High aplica el algoritmo de smith watterman para calcular la matriz con los scores máximos.
Calcula las posiciones que se evaluarán, recuerda que la posición que se prueba se evalúa contra la diagonal el valor superior y el valor a la izquierda: Valor de la diagonal diag = H[i-1][j-1] + match(s1[i-1], s2[j-1]) Valor de posición superior up = H[i][j-1] - 1 Valor de posición izquierda left = H[i-1][j] - 1 Calcula el valor máximo de las posiciones que se comparan H[i][j] = max(0,left, up, diag)
Llena la matriz Traceback ó T para poblar los valores que se compararán y reasigna el valor correcto a la posición de la matriz High o H if H[i][j] == 0: T[i][j] = 0 if H[i][j] == left: T[i][j] = 1 if H[i][j] == up: T[i][j] = 2 if H[i][j] == diag: T[i][j] = 3 if H[i][j] >= max_score: max_i = i #guardar este valor para realizar el Traceback max_j = j #guardar este valor para realizar el Traceback max_score = H[i][j]