¡Descarga Validación de paracetamol y más Monografías, Ensayos en PDF de Química solo en Docsity!
“VALIDACIÓN DE LA METODOLOGÍA ANALÍTICA
DE PARACETAMOL Y APLICACIÓN A
UN ESTUDIO DE BIOEXENCIÓN”
Supervisor de Práctica
Prof. Q.F. Edda Costa C. Departamento de Ciencias y Tecnología Farmacéutica
Monitor de Práctica
Q.F. Karla Rodríguez R. Jefe de Control de Calidad Prod. Farmacéuticos Medipharm®
Unidad de Práctica Tutorial para optar al título de Químico Farmacéutico
FRANCISCO JAVIER GAETE CASTRO Santiago
ÍNDICE
- Índice ………………………………………………………………………………….……
- Resumen ………………………………………………………………………….….……
- Descripción del laboratorio ……………………………………………………….……
- Introducción ……………………………………………………………………………....
- Conceptos …………………...…………………………………………………...…..…...
- Objetivos …………………………………………………………………………………
- Marco teórico ……………………………………………………………………………
- Materiales …………………………………………….………………………………….
- Metodología ……………………………………………………………………………..
- Parámetros para validar ………………………………………………………….……
- Resultados y discusiones …………………………………………………….…..…..
- Perfiles comparativos ……………………………………………………………...….
- Conclusiones ………………………………………………………………………...…
- Bibliografía .…………………………………………………………………………...…
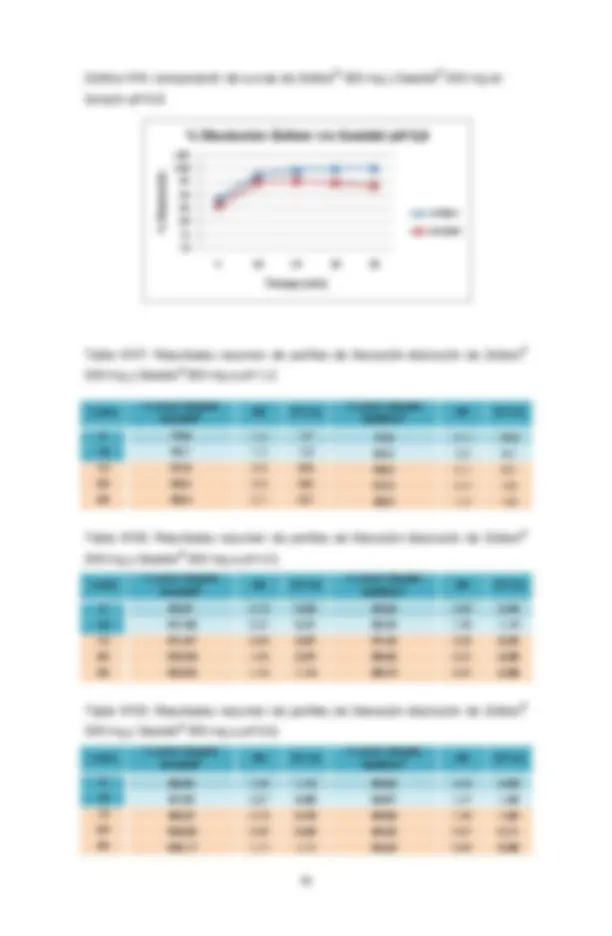
cotejados para llevar a cabo la comparación de las curvas, y eventualmente realizar el cálculo del factor de similitud f2 , que en este caso no es necesario de realizar debido a la muy rápida liberación del activo (mayor al 85% de la cantidad declarada antes de 15 minutos), lo que demuestra que la liberación no constituye un obstáculo para la posterior absorción y que este tiempo depende en forma exclusiva del vaciamiento gástrico.
DESCRIPCIÓN DEL LABORATORIO Y
CENTRO BIOFARMACÉUTICO
Productos Farmacéuticos Medipharm®^ es un laboratorio de productos farmacéuticos perteneciente al consorcio de empresas Salcobrand®. Actualmente funciona como una droguería debido a que no posee planta productiva propia, pero ejerce las labores de almacenamiento y distribución de productos terminados en cuarentena y liberados por diferentes “maquiladores”. Actualmente posee tres departamentos, los cuales son:
Departamento de Desarrollo: encargado de formular, modificar y escalar la fabricación de productos farmacéuticos, tanto productos nuevos como los ya existentes en el mercado.
Departamento de Control de Calidad y Centro de Bioequivalencia: está encargado de realizar estudios de estabilidad ambiente y acelerado, control de calidad de rutina de los productos fabricados que se encuentran en cuarentena por los laboratorios maquiladores, como también los productos que ya se encuentran liberados para su distribución en farmacias. Actualmente el laboratorio de control de calidad de Medipharm®^ está autorizado por el ISP para realizar estudios in vitro para optar a bioexención [9]. Dentro de los análisis realizados en dicho laboratorio se puede mencionar:
Medición de pH y valoraciones potenciométricas. Valoraciones volumétricas. Análisis por cromatografía líquida de alta eficiencia (HPLC) Análisis por espectrofotometría ultravioleta (UV). Análisis por espectrofotometría infrarroja (IR). Ensayos de cinética de disolución. Determinación de humedad por método de Karl Fischer. Ensayo de dureza de comprimidos. Ensayo de desintegración. Ensayo de friabilidad. Ensayo de viscosidad.. Estudios de bioexención. Estudios de solubilidad de principios activos.
Departamento de Validaciones : es el encargado de diseñar, corregir y mejorar las metodologías analíticas de productos nuevos y ya comercializados, además de realizar las tareas de documentar todos los procesos y antecedentes para la autoridad sanitaria.
Se entiende la bioequivalencia como un estándar de calidad para medicamentos de múltiples orígenes, impulsado principalmente desde los gobiernos de países desarrollados donde el costo derivado de medicamentos y de los sistemas de salud cada año se incrementa cada vez más, resultando una buena forma de acceder a medicamentos de forma más asequible y sin menoscabar la calidad de éstos [3, 4].
Para considerar a un medicamento similar como equivalente terapéutico de otro, el cual es un innovador, deben establecerse varias cualidades como:
Equivalente Farmacéutico : debe tener mismo principio activo, misma potencia, misma dosis y forma de administración, etiquetado y rotulado comparables. Bioequivalencia : contempla mediciones in vivo de fracciones del principio activo en fluidos biológicos, comparaciones farmacodinámicas in vivo , comparaciones clínicas in vivo , comparaciones in vitro. Se establece que la equivalencia terapéutica de una formulación es la suma de su calidad de equivalente farmacéutico más la bioequivalencia [4].
Juntos con las soluciones, también aparecen las dificultades y entre ellas, la más compleja de sortear surgió con las primeras exigencias de la autoridad regulatoria respecto de los estudios de bioequivalencia, fue que éstos resultaron ser muy costosos, debían ser realizados en centros especializados tales como hospitales, clínicas y centros médicos o de investigación, requerían de muchos voluntarios y que éstos fueran de un perfil biológico homogéneo, en cuanto a edad, sexo, estilo de vida, enfermedades existentes, etc., lo cual retrasó considerablemente los plazos de implementación y desalentó a la industria farmacéutica a realizar dichos estudios, especialmente en los países en vías de desarrollo como el nuestro.
Ello impulsó una alternativa de estudio que permite asegurar la intercambiabilidad de medicamentos a través de estudios in vitro , y que en general tienen buena correlación con lo que acontece en el organismo: los estudios de liberación-disolución in vitro para optar a bioexención de estudios de bioequivalencia in vivo con el fin de establecer equivalencia terapéutica.
El estudio de bioexención se puede definir como un estudio alternativo al estudio de bioequivalencia in vivo mediante la demostración de equivalencia terapéutica in vitro para un cierto grupo de fármacos que cumplen los requisitos señalados por el Sistema de Clasificación Biofarmacéutica (SCB) [3,5]. Dicho
estudio consiste básicamente en la comparación de los perfiles de disolución in vitro entre el producto farmacéutico innovador y el producto similar. Para realizar dichos estudios se deben cumplir condiciones básicas tales como:
Los productos farmacéuticos deben ser fabricados bajo las Buenas Prácticas de Manufactura, en inglés Good Manufacturing Practices GMP, además de cumplir las especificaciones de calidad de las farmacopeas oficiales. Los principios activos deben tener suficientes antecedentes bibliográficos que demuestren su buena permeabilidad y solubilidad en distintos pH fisiológicos, datos que se encuentran disponibles en la literatura especializada a través del SCB, planteado por Amidon et. al, sistema avalado y respaldado por la Organización Mundial de la Salud (OMS) [5,6].
Actualmente, varios autores están desarrollando sistemas de clasificación alternativos al SCB, con la idea de incorporar otros aspectos que son relevantes para el comportamiento farmacocinético de una formulación farmacéutica. Un ejemplo de sistemas alternativos es el SCBDF planteada por Benet et al ., el cual se utilizará como referente en esta investigación [7,13].
Las agencias regulatorias europea y estadounidense (EMEA y FDA) han adoptado los criterios del SCB para establecer equivalencia terapéutica entre formulaciones de un mismo activo; para el caso especial de los fármacos Clase I del sistema SCB (altamente solubles y altamente permeables) no es necesaria la demostración comparativa de bioequivalencia in vitro siempre y cuando los excipientes usados y el proceso de manufactura no afecten la absorción del activo y que el principio activo no posea estrecho margen terapéutico. Actualmente también se extiende la posibilidad de realizar estudios de bioexención a fármacos de clase III del SCB (alta solubilidad y baja permeabilidad) [8].
Es así que se resuelve de una manera sencilla, económica y rápida la necesidad de contar con estudios de bioequivalencia en los países en vías de desarrollo como Chile, promoviendo metodologías avaladas internacionalmente por investigadores expertos en la materia, realizables con instrumental disponible en la mayoría de los laboratorios nacionales, los cuales poseen facilidades de mantención y calificaciones periódicas y a un costo económico y de tiempo mucho más acotado. A pesar de todos los beneficios mencionados anteriormente, el mayor impacto se relaciona con la mejora en la accesibilidad de los pacientes a los medicamentos sin extender el plazo de los estudios.
Equivalentes terapéuticos [5]:
Se le denomina así a productos farmacéuticos equivalentes terapéuticos si son además equivalentes farmacéuticos y, después de la administración en la misma dosis molar, sus efectos, con respecto a eficacia y seguridad, son esencialmente los mismos, determinados por estudios apropiados (clínicos, farmacodinámicos, de bioequivalencia, o " in vitro "). Tales productos deben estar adecuadamente rotulados y ser manufacturados cumpliendo con las normas vigentes de Buenas Prácticas de Manufactura (GMP).
Bioexención [5] :
Es la prerrogativa de la autoridad regulatoria para eximir de la obligación de tener que presentar estudios in vivo para el establecimiento de la equivalencia terapéutica, la cual puede demostrarse mediante estudios in vitro. En resumen es un estudio científico acotado y alternativo a los estadios de bioequivalencia in vivo a través de la demostración de equivalencia terapéutica in vitro. Está limitada a un determinado grupo de fármacos, los cuales deben cumplir como condición intrínseca ser muy solubles y muy permeables. Generalmente se basa en los datos proporcionados por el SCB.
OBJETIVOS
OBJETIVO GENERAL
Validar la metodología analítica para realizar el estudio de bioexención de Gesidol®^ comprimidos 500 mg.
OBJETIVOS ESPECÍFICOS
Conocer y llevar a cabo los distintos análisis realizados en el laboratorio de control de calidad de Medipharm®, tanto los realizados en forma rutinaria, como los destinados a resolver situaciones concretas, usando de referencia las farmacopeas oficiales así como también las metodologías internas del laboratorio.
Realizar una recopilación bibliográfica sobre paracetamol en la que se justifique debidamente la razón por la cual este principio activo es apto para optar a la bioexención de estudios de bioequivalencia in vivo.
Llevar a cabo la validación de la metodología analítica para Gesidol®^ 500 mg involucrando los parámetros exigidos por la autoridad sanitaria tales como: linealidad, precisión, exactitud, rango, especificidad, estabilidad, robustez, influencia del sistema de filtración y precisión intermedia.
Obtener los perfiles de liberación del medicamento innovador y de Gesidol® 500 mg y verificar si son comparables mediante el factor de similitud f 2.
El porcentaje de la dosis absorbida se puede calcular sumando el porcentaje biotransformado a través del hígado mediante metabolismo de primer paso, a la biodisponibilidad absoluta. Esto sugiere que la fracción de dosis absorbida es mayor que 80% siendo que el valor límite de un principio activo para ser clasificado como muy permeable por los actuales criterios del SCB es una fracción de la dosis absorbida mayor al 90% (según la FDA). Estos datos llevan a la conclusión de que paracetamol sea clasificado como poco permeable.
El SCB obtiene los datos de solubilidad mediante determinaciones analíticas y los datos de permeabilidad son obtenidos a través de ensayos de difusión a través de membranas biológicas. La Tabla N°1 hace referencia clasificación de paracetamol a través del sistema SCB propuesto por Amidon et. al.:
Tabla Nº1: Clasificación de Paracetamol bajo los criterios de la SCB [12]:
Dentro de esta clasificación surge una de las primeras dificultades para llevar adelante el estudio. Según los datos de solubilidad y permeabilidad señalados en el SCB, paracetamol aparece catalogado como clase IV y según otros autores, es clasificado como clase III teniendo siempre como limitante entre ambas clases a la permeablidad, por lo cual no es apto para ser sujeto de un estudio de bioexención. Si bien se tiene que reconocer que el SCB ha permitido ahorrar mucho tiempo y costos en poder establecer la necesidad o no de realizar estudios in vivo , también es cierto que omite importantes detalles que modifican la biodisponibilidad de un principio activo y que, necesariamente deben ser tomados en cuenta a la hora de realizar un estudio de bioexención. Entre las omisiones que se deberían tomar en cuenta por parte del SCB respecto de los fármacos a estudiar son:
Molécula: Paracetamol (Acetaminofeno) Número CAS: 103 - 90 - 2 Categoría: Agentes que influyen en el sistema nervioso central Subcategoría: Analgésicos (otros) Fórmula química: C 8 H 9 NO 2 Peso molecular: 151.16 g/mol pKa: 9. Solubilidad mínima (mg/mL): 0. Permeabilidad en humanos (x 10-^4 cm/s): N/A logP: 0.55 Baja permeabilidad Lista de países: Dosis mínima (mg) Dosis máxima (mg) Solubilidad Clase SCB (logP) EEUU (^) 300.0 750.0 Baja Clase IV OMS (^) 100.0 500.0 Baja Clase IV Japón (^) 200.0 300.0 Baja Clase IV Surcorea (^) 80.0 500.0 Baja Clase IV Reino Unido 250.0^ 1000.0^ Baja Clase IV
Tipo y cuantía de la metabolización. Efecto de los transportadores de membrana. Influencia de los alimentos. Principales rutas de eliminación.
Surge un sistema de clasificación complementario y claramente no excluyente del SCB como herramienta para resolver de buena manera estas variables no contempladas. Es el Sistema de Clasificación Biofarmacéutica basada en la Disposición del Fármaco o droga, SCBDF (en inglés BDDCS) [7,13]. Este sistema fue propuesto por los profesores Chu-Yuan Wu y Leslie Z. Benet de la Universidad de California - San Francisco el año 2005 y hasta el momento ha gozado de gran aceptación dentro de la comunidad científica ya que correlaciona de mejor manera el comportamiento de un principio activo in vitro/in vivo.
Sistema de Clasificación Biofarmacéutica basada en la Disposición del Fármaco (SCBDF) [7]
La diferencia fundamental entre el SCB y el SCBDF es el reemplazo de la variable “ permeabilidad” utilizada por el SCB por la variable “metabolización” (expresado como porcentaje), postulando que la cuantía de la metabolización de un principio activo es directamente proporcional a la absorción que éste experimenta en el organismo[9]. Wu y Benet afirman que clasificar a los principios activos por su porcentaje de metabolización es menos susceptible de error que hacerlo considerando la permeabilidad, proponiendo al SCBDF como reemplazo de esta variable dentro de los estudios de bioexención [7,13].
Wu y Benet sugirieron que un fármaco cuya principal vía de eliminación ocurre través de procesos metabólicos, en general exhibía una alta permeabilidad, y por el contrario, los fármacos con eliminación inalterada, ya sea renal o biliar, se clasificaron como de baja permeabilidad. Los autores definieron “ altamente metabolizado ” a los principios activos administrados por vía oral cuya tasa de recuperación de metabolitos es mayor al 70% y los “escasamente metabolizados” con una recuperación inferior al 50%. De esta manera surgen las nuevas clasificaciones basadas en el SCBDF [12,13]. En la Tabla N°2 se hace referencia a los criterios utilizados por el sistema SCBDF:
Tabla Nº3: Extracto de datos de disposición de algunos activos pertenecientes a la lista de medicamentos esenciales de la OMS reclasificados al sistema SCBDF [7,15].
Es notable la diferencia que existe entre la clasificación que tiene paracetamol bajo el SCB (clase IV) y bajo el SCBDF (Clase I) lo que se puede explicar por las siguientes consideraciones [14]:
Bajo el SCBDF, el lumen intestinal es lo suficientemente poroso y permeable para permitir rápidamente el paso de moléculas de bajo peso molecular, no polares y solubles. Además, la alta permeabilidad y solubilidad de los compuestos clase I permiten saturar los transportadores de recaptación y las bombas de eflujo de los cuales son sustratos, aunque esto no tiene importancia clínica pues no constituyen elementos limitantes.
Bajo el SCBDF se establece que los alimentos ricos en grasas no tienen un efecto significativo en los fármacos clase I porque la influencia de los transportadores en la biodisponibilidad es mínima para estas moléculas. Lo que sí alteran es el tiempo máximo debido a que los alimentos ricos en grasas tienden a retardar el vaciamiento gástrico y por ende, retardan el inicio de acción de algunos principios activos.
Al ser metabolizados por el hígado y el lumen intestinal, la interacción ocurre solamente en los transportadores de membrana.
Es así que usando estos nuevos criterios, el SCBDF extiende el número de fármacos Clase I que pueden optar a estudios de bioexención, permitiendo en varios casos que algunos fármacos clasificados como clase III según el SCB y que no eran aptos para estudios de bioexención, fueran reclasificados como Clase I bajo el SCBDF [7,14]. En general, las conclusiones obtenidas de este enfoque, permiten deducir que los fármacos altamente permeables pero escasamente
Nombre Principio Activo
Dosis (mg)
Solubilidad (mg/mL)
Biodisponibilidad (%)
Grado de metabolización SCB^ SCBDF Ácido Acetilsalicílico 500 4,6 Sin datos Alto 3 1 Clomifeno Citrato^50 1 90 Bajo^1 Levotiroxina Sódica 0,1^ 0,15^70 Bajo^1 Metildopa^250 10 25 Alto^3 Paracetamol[14]^500 0,1^75 Alto^4 Praziquantel 600 0,4 80 Alto 2 2 Salbutamol^4 33 44 Bajo^3 Sulfasalazina^500 0,01^ No aplica^ Alto^2
metabolizados son sustancias polares de bajo peso molecular y son absorbidas mayoritariamente por mecanismos de transporte activo. A su vez se puede concluir que la biodisponibilidad es alta (mayor al 90%) o completa cuando el principio activo experimenta un gran metabolismo (mayor al 90%) [14].
En general, para obtener buenas correlaciones in vivo / in vitro , se deben tener en cuenta varios factores, entre ellos, la pérdida del principio activo por factores enzimáticos (metabolización en el tracto gastrointestinal por enzimas o flora intestinal), factores no enzimáticos (hidrólisis), etc. En la Tabla N°4 se hace referencia a las principales diferencias entre los sistemas SCB y SCBDF:
Tabla Nº4: Diferencias entre los sistemas SCB y SCBDF [7]. SCB SCBDF Criterios de clasificación
Solubilidad. Permeabilidad.
Solubilidad. Metabolización. Correlación Más compleja in vivo/in vitro. Menos compleja in vivo/in vitro. Activos admisibles de Bioexención Número relativamente reducido.^
Número mayor de activos respecto al SCB. Influencias externas (^) Influencia de tInfluencia de aransportadoreslimentos.. No tienen influencia los transportadores^ No tienen influencia los alimentos..
Es así que la sumatoria de los criterios utilizados oficialmente por las principales agencias regulatorias de medicamentos a nivel mundial (FDA y EMA) y el nuevo criterio de la SCBDF, se postuló ante la autoridad sanitaria que paracetamol es un fármaco apto para postular al estudio de bioexención, lo cual resultó ser aceptado mediante resolución exenta emitida por el Subdepartamento de Biofarmacia del ISP, la que fue recibida en el laboratorio de control de calidad [16].
METODOLOGÍA
La validación de la metodología analítica y la obtención de los perfiles de disolución de los productos farmacéuticos innovador y similar, se realizarán de acuerdo a lo estipulado en la guía técnica G-BIOF 2[8]^ redactada por el ISP, la cual establece los parámetros mínimos que se deben determinar en el proceso de validación. Esta guía recomienda y especifica variados aspectos como por ejemplo lo es la técnica de análisis; se puede utilizar espectrofotometría UV-visible por ser un método sencillo y con un mínimo gasto de solventes aunque, en este caso se prefirió utilizar cromatografía HPLC por ser una técnica que permite la automatización del proceso de lectura de numerosas muestras, permite detectar más fácilmente las eventuales interferencias de algunos excipientes y posibles productos de degradación, además de ser un método más sensible y con un menor límite de cuantificación.
Producto en estudio:
Principio activo: Paracetamol Forma farmacéutica: Comprimidos Nombre comercial: Gesidol®^ comprimidos 500 mg
Producto de referencia o comparador:
Principio activo: Paracetamol Forma Farmacéutica: Comprimidos Nombre comercial: Zolben®^ comprimidos 500 mg
MÉTODO DE DISOLUCIÓN [5]
Es el ensayo in vitro que da cuenta de forma experimental el comportamiento de dos formulaciones en condiciones ambientales idénticas, de manera de poder establecer los perfiles cinéticos de disolución de un principio activo desde una forma farmacéutica sólida con la finalidad de compararlos mutuamente.
Los métodos de disolución más comúnmente empleados son los del canastillo (aparato 1) y de la paleta (aparato 2) de la USP. Estos métodos son suficientemente flexibles para permitir evaluar las características de disolución de una gran variedad de productos, por lo que se recomienda su uso, a menos que se demuestre que no son satisfactorios. Los aparatos para probar la disolución utilizados en esta evaluación deberán conformarse a los requisitos de la USP.
CONDICIONES DEL ENSAYO DE DISOLUCIÓN
Aparato II de la USP (paletas). Velocidad: 75 rpm. Número de unidades: 12 comprimidos (un lote). Tiempos de muestreo: 5, 10, 15, 20, 30 minutos. Temperatura: 37°C ± 0,5°C. Volumen medio: 900 mL de los medios detallados posteriormente (soluciones amortiguadoras de pH 1,2; 4,5 y 6,8). Los tiempos de muestreo, la frecuencia y el tiempo total del ensayo, tanto del producto de prueba como el de referencia deben permitir la obtención de un perfil adecuado de disolución para poder aplicar los criterios de similitud. Los tiempos de muestreo deben ser exactamente los mismos para ambos productos. En el caso de paracetamol, es una forma farmacéutica que presenta una rápida disolución en la formulación que se pretende estudiar, lo cual fue evaluado previamente a través de una prueba de disolución sobre 3 comprimidos. Se decide tomar muestras a los 5, 10, 15, 20 y 30 minutos, considerando este último tiempo como punto sobre el cual se encuentra al menos 85% disuelto. Si bien es cierto que paracetamol está clasificado en Clase IV en el SCB, la información anterior permite establecer que la solubilidad en los pH de prueba no sería una limitante en la solubilidad/disolución del fármaco.
Para establecer similitud o diferencia entre las curvas del medicamento innovador y del medicamento ensayado, el cálculo de factor de similitud se realizará a cada uno de los tiempos indicados en el punto anterior, incluyendo el tiempo por sobre el 85% disuelto. Si se obtiene un porcentaje mayor al 85% disuelto a los 15 minutos o menos, en los 3 medios evaluados, se considera a las formulaciones como formas farmacéuticas de liberación muy rápida y, por lo tanto no será necesaria la comparación de perfiles con un cálculo de f2. Esto se debe a que la Norma Chilena referida a equivalencia terapéutica establece que cumpliéndose esta condición, no es necesaria la comparación de los perfiles, aunque esto no exime la presentación de los perfiles de disolución de los productos en estudio y del producto de referencia[3,5].
Soluciones amortiguadoras [17]:
En el caso de los tampones, se prepararan idénticamente a lo establecido en la USP 36, capítulo <711> “soluciones”, tomando 900 mL de cada solución amortiguadora previamente desaireada y calentado a 37°± 0,5°C los cuales son los siguientes: