Scarica Bioinformatica - prof. Percudani e più Appunti in PDF di Bioinformatica solo su Docsity!
Bioinformatica Studio dei problemi biologici attraverso le metodologie dell’informatica.Sinonimi: biochimica computazionale, biologia molecolare computazionale Viceversa Biocomputazione, algoritmi genetici, reti neurali metodi informatici di applicazione generale che si ispirano ai principi della biologia. Scopi della bioinformatica: ● Gestione dei dati biologici: mantenimento, organizzazione, distribuzione... ● Analisi dei dati biologici : inferenze e predizioni sul significato biologico Gli oggetti di studio della bioinformatica sono:
_- sequenze di acidi nucleici
- sequenze di proteine
- strutture di macromolecole_ Il formato FASTA è un formato in cui possono essere presentate ed utilizzate le sequenze di DNA e proteine.
Nelle banche dati è rappresentato solo il filamento 5’-3’. Il codice identificativo della sequenza inizia con “>” ( es. >38F13B → tale codice rappresenta l’ACCESSION NUMBER che non è altro che il numero identificativo di ciascun acido/proteina). L’ordine di grandezza del genoma umano è 10^9 , in particolare 3 x 10^9 bp e il numero di geni è di 20- 30 mila codificanti per proteine. Qui di seguito è rappresentato uno schema che deriva dal dogma centrale della biologia: Gli organismi modello maggiormente utilizzati sono: ratto, topo, Drosophila Melanogaster (fruitfly), Aradopsis Thaliana (pianta), Xenoupus Laevis (rana), Sea urchin (riccio di mare), Saccharomyces Cerevisiae, E. Coli, Caenorhabtidis elegans (nematode). Le 3 grandi famiglie Archea, Bacteria e Eukarya derivano da un ancestore universale comune. Esiste un trasferimento orizzontale in cui vediamo il collegamento tra mitocondrio/cloroplasto e i batteri. Da tenere presente sia la discendenza darwiniana, sia quella orizzontale, entrambe utili per classificare proteine e geni.
**Di seguito i 2 principali approcci/sistemi utilizzati in bioinformatica:
- Interfaccia grafica basata su browser come Chrome
- Command Line basata su Linux:** funzionano dando comandi scritti, servono per l’analisi in larga scala quando si lavora con grandi quantità di dati da elaborare. Ecco i principali programmi usati:
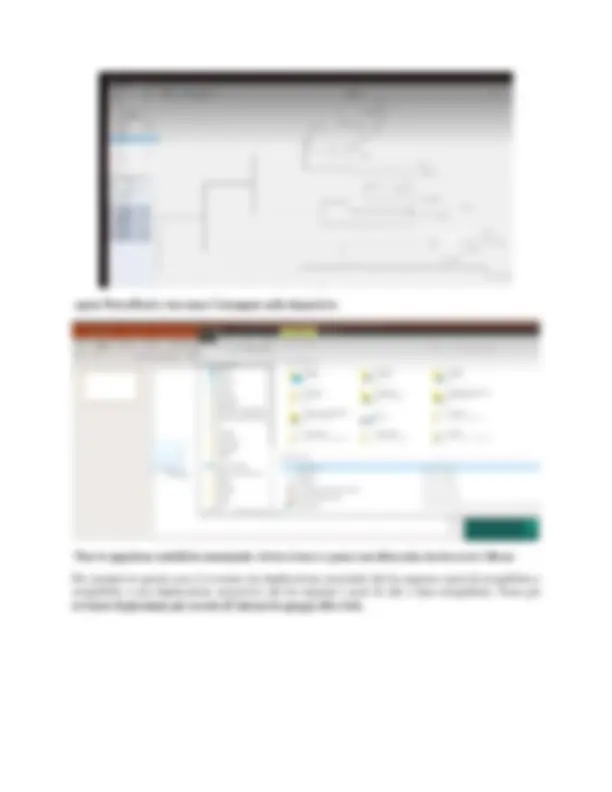
BIOINFORMATICA Lezione 2- 29.09. Su Elly2021 di “bioinformatica” nella sezione annunci è presente il link alla pagina degli esercizi, in cui è presente un link, che una volta aperto mostra: esercizi guidati, materiale didattico ed eventuali articoli extra. Prima cosa da fare oggi è accedere ad un server Linux attraverso il client SSH, che permetterà di dare istruzioni con la riga di comando. Il software utilizzato è PuTTY, che è scaricabile dal seguente link: https://www.putty.org/. Invece, la guida per FortiClient per connettersi da casa alla rete UniPr è al seguente link: https://noc.unipr.it/risorse/public/vpn/forticlient/guide/Installazione_FortiClient_Windows.pdf. Il download del client è fondamentale, per poter utilizzare il server Linux con il proprio computer, perché permette di accedere ai server dell’università. PuTTY è il client da utilizzare per il collegamento ed una volta scaricato ed aperto apparirà la seguente schermata: L’indirizzo del server è didattica-linux.unipr.it (si trova nel primo esercizio) e viene inserito nella casella indicata da “ Host Name ” (vedi immagine precedente). La “ Connection type ” è SSH, che consente di comunicare con un terminale (non è presente finestra grafica ma solo finestra di comando). Una volta inseriti questi dati clicchiamo su “Open” ed apparirà la seguente finestra (accade solo la prima volta che lo apriamo): Quindi cliccare “Sì”.
Per la creazione di una directory si utilizza il comando: mkdir bioinfo (spazio dopo mkdir). La directory è una “carpetta” (=cartella del pc), quindi un contenitore di file. Per cui il comando “mkdir” serve per creare uno spazio separato dal resto della home directory (N.B il comando e l’oggetto del comando devono essere separati da uno spazio, perché lo spazio ha uno significato ben preciso in Linux. Se il comando va a buon fine non si ha nessuna indicazione, al contrario si ha un qualche “segnale”.) A questo punto se usiamo il comando “ls” avremo tra le nostre directory “bioinfo” Per accedere ad una particolare “carpetta” (directory), come per esempio quella appena creata “bioinfo”, il comando è cd bioinfo (spazio dopo cd). Come “prompt” si ottiene “bioinfo” (vedi cerchiato in rosso): A questo punto con il comando “ls” non si vede nulla, ma con il comando “ls - a” si vedono due “carpette” speciali che sono indicate
da:. che indica la directory corrente, mentre i.. (due punti)
indicano la directory precedente e consentono di tornare indietro. (cerchiati in rosso in figura a destra). Infatti per tornare nella directory precedente si usa il comando cd .. (cd spazio e due punti successivi). L’esercizio (primo esercizio presente sulla pagina esercizi nella sezione annunci di Elly) da svolgere prevede la creazione di un file contente sequenze FASTA, che vengono inserite come file di testo nella directory bioinfo. Come creare un file con sequenze FASTA Prima di tutti ci colleghiamo nella banca dati NCBI per recuperare le sequenze. Nello specifico saranno necessarie delle sequenze “refseq” su gene di NCBI.
- Aprire NCBI Gene
- Cercare la sequenza proteica dell’emoglobina beta delle cinque specie indicate nell’esercizio → Scriviamo il nome del gene HBB + il nome della specie. Per esempio: HBB Homo sapiens (Attenzione nella ricerca in banche dati, non sempre il primo record è quello d’interesse)
- Clicchiamo su HBB – hemoglobin subunit beta
- Prendiamo la sequenza proteica corrispondente: scorrere nella pagina fino a quando non si arriva qui e cliccare su “ Go to reference sequence details ”:
- Troviamo, a livello di “ reference sequence details ”, le sequenze di riferimento: del gene, del mRNA e della proteina. La sequenza del gene si trova nella sezione “genomica”, mentre nella sezione “ mRNA and Protein(s) ” è presente sia la sequenza del mRNA che della proteina. La sequenza della proteina inizia sempre per “N P o X P ”.
- Cliccare a livello dell’ accession number per la sequenza della proteina, in questo caso: NP_000509.
- Otteniamo una pagina in cui nella parte finale è presente la sequenza proteica in formato GeneBank e tutto ciò che la precede è il metadato cioè l’annotazione al dato ( dato=sequenza della proteina). Per poter utilizzare la sequenza, il formato GeneBank non è molto pratico perché quasi tutti i programmi non saranno in grado di interpretarlo mentre tutti i programmi sono in grado di comprendere il formato FASTA.
- Recuperiamo la sequenza in formato FASTA andando a cliccare sulla dicitura “FASTA” presente subito dopo il nome della proteina
- Otteniamo la sequenza in formato FASTA: In cui all’inizio è presente il “>” che indica l’inizio del record, successivamente l’ Accession Number , poi una descrizione (hemoglobin subunit beta) e l’organismo tra parentesi ed infine la sequenza in formato FASTA data da una successione di lettere. Il formato FASTA è abbastanza permissivo per cui si può andare a capo senza problemi oppure inserire spazi, però chiaramente non posso inserire lettere che fanno parte della sequenza altrimenti si potrebbe confondere il sistema.
- Copio la sequenza a partire dal maggiore e la incollo in un file di testo. L’editor di testo da poter utilizzare su Windows è Notepad ++ (non Blocco note perché troppo basico) Questi passaggi si ripetono per tutte le sequenze necessarie indicate nell’esercizio.
- Salvare il file e indicare come estensione “ All types (.)** ”, per non avere un’estensione automatica. Per poter decidere l’estensione bisogna cliccare sulla tendina affianco “Salva come”. Il nome del file è “HBB.fa”(N.B alle maiuscole e minuscole perché per Linux è presente una grande differenza)
- Prima di effettuare il trasferimento, a livello delle impostazioni di trasferimento (cerchiato in rosso in immagine precedente) scegliere “testo”
- Per visualizzarlo su Linus, dopo aver aperto l’interfaccia, inserisco il comando: ls (comando per mostrare ciò è presente nella directory corrente). Questo permette di visualizzare la presenza del file “HBB.fa” all’interno di bioinfo:
- Per visualizzare il contenuto del file HBB.fa, uso il comando: cat HBB.fa. In questo modo vedo le due sequenze precedentemente aggiunte: L’esercizio prevedeva di cercare molte più di sequenze, ma le restanti erano già state preparate dal professore. Dunque, per poterle trasferire (dalla directory del professore chiamata “tmp” alla nostra) usiamo il comando: cp /tmp/HBB.fa. (N.B: dopo “cp” è presente uno spazio e anche dopo HBB.fa). Quindi così abbiamo sovrascritto il file HBB.fa precedente con questo nuovo e così otteniamo un file con tutte le sequenze. Se utilizziamo il comando cat HBB.fa possiamo vedere che il file presenta tutte le sequenze richieste dall’esercizio. Ora facciamo la stessa cosa per HBA, andando a copiare nuovamente il file dalla directory del professore, usando il comando: cp /tmp/HBA.fa. Se utilizziamo ora il comando ls , si vedranno i due file HBB.fa e HBA.fa : Per vedere il contenuto dei due file insieme, uso il comando cat HB?.fa → In questo modo vedo i due file insieme uno dopo l’altro. Sarebbe stato possibile, allo stesso modo, utilizzare il comando cat e il nome dei file successivamente, senza l’utilizzo del carattere speciale “?”.
Il comando grep serve per trovare delle “cose” in un file o su più file. Per esempio, possiamo sfruttare il comando grep Homo HBA.fa per cercare la riga di intestazione della sequenza umana nel file HBA.fa. Per ricercare sia nel file HBA.fa che nel file HBB.fa utilizzo il comando: grep Homo HB?.fa
Per quanto riguarda la porzione in blu, ossia quella predetta bioinformaticamante, avete un’idea di quale sarà il sistema che mi consente di associare una probabile funzione a queste sequenze? L’omologia è il sistema che consente l’annotazione della maggior parte delle proteine che non sono direttamente studiate (trovo un rapporto di omologia con qualche proteina studiata, quindi faccio una sorta di trasferimento di funzione). Seppur questo grafico identifichi la % di proteine batteriche, stesso discorso vale per quelle eucariotiche. Qui riportata è la statistica di UniProt: la linea verde identifica il numero delle proteine che sono annotate manualmente sia su evidenze sperimentali sia su evidenze bioinformatiche supervisionate da un esperto; in blu viene riportato il numero delle proteine che sono annotate automaticamente dal sistema grazie un algoritmo del computer in cui non c’è la supervisione di un esperto. La maggior parte delle proteine sono annotate per omologia con proteine note, per cui si trova una somiglianza significativa 2 proteine distinte erano in origine un’unica entità e se conosco la funzione per via diretta di una delle 2, allora posso predire la funzione dell’altra. Può essere un’operazione banale o molto difficile in alcuni casi. Il caso banale è quando avete un’evidenza così forte e la garanzia che si tratti dello stesso gene rispetto al gene di riferimento che è stato studiato, che siete abbastanza sicuri di trasferire la funzione. Nel nostro caso in slide non vi sono trattini (-) che indicano che c’è stata una delezione perché sono proteine talmente conservate che in nessuna di queste è avvenuta una inserzione/delezione di amminoacido e sono avvenute anche pochissime mutazioni (sottolineature in rosa). Abbiamo altri casi in cui non è così semplice assegnare la funzione sulla base dell’omologia:
- posso trovare somiglianza con proteine a funzione nota, ma non in modo conclusivo; per esempio, posso non essere sicuro perché in caso di paralogia (geni che derivano da un evento di duplicazione genica) aumenta la possibilità che la funzione sia cambiata nel tempo
- posso trovare proteine simili in banca dati, ma a funzione ignota (nessuna delle proteine che ho trovato è stata mai studiata e determinata come funzione sperimentalmente)
- posso trovare che la proteina non assomigli a niente (casi sempre più rari ma possibili).
Anche in presenza di molte evidenze di tipo bioinformatico/ molti dati riguardanti l’omologia e la struttura, può non essere facile identificare e predire la funzione della proteina. Un esempio che vi riporto è la proteina PROSC, il dominio è conosciuto come DUF0001: domain of unknown function. È una proteina presente in tutti gli organismi con pochissime eccezioni, assomiglia a proteine con ruolo di racemizzazione dell’alanina, la struttura è nota, conosciamo il cofattore, conosciamo anche il fenotipo del deleto, ma non sappiamo ancora ad oggi la funzione. In altri casi è possibile che l’analisi bioinformatica riesca a dare indicazioni sul funzionamento della proteina. In generale la cosa che può aiutarmi è trovare l’evidenza di un’associazione funzionale tra la mia proteina X d’interesse e altre proteine, nella speranza che alcune di queste siano caratterizzate funzionalmente e possano darmi delle informazioni sul funzionamento di X trovo delle associazioni funzionali (non un rapporto di omologia), trovo qualcosa che unisca il funzionamento di X e le altre proteine (A / B / C). Cosa significa dal punto di vista biologico che la funzione di una proteina è associata alla funzione di un’altra proteina? Possono formare un complesso macromolecolare e quindi quella proteina è associata fisicamente all’altra/ possono essere enzimi della stessa via metabolica/ possono far parte di una via di trasduzione del segnale ecc quindi ci sono tantissimi casi in cui la proteina funziona in associazione ad altre. Come facciamo in bioinformatica a predire questa associazione funzionale tra geni? Ci sono software che ci fanno proprio vedere delle evidenze di associazione tra proteine diverse; queste evidenze di associazione derivano da dati di tipo diverso. Uno dei software per fare questo tipo di analisi bioinformatica si chiama STRING, utilissimo per trovare evidenze di associazioni e poter fare predizioni.
Per fare un esempio per come può essere utilizzata e di come si può ragionare in presenza di queste evidenze, vi faccio vedere come è stato scoperto il 22esimo amminoacido ossia la Pirrolisina; deriva dalla Lys, presenta un gruppo pirrolico, è utilizzato solo da alcuni batteri ed è codificato da un codone di stop che è stato riassegnato. La primissima evidenza per la scoperta di questo aa è stata che una particolare proteina (metilammina metiltransferasi) al sequenziamento mostrava un codone di stop, mentre invece l’organismo si sapeva dovesse produrre una proteina funzionante. Sequenziando vicino a questi 2 geni che contengono questo putativo codone per un diverso aa, si è visto che c’era 1 gene che codificava per un tRNA particolare con anticodone CUA. L’anticodone CUA legge UAG, dunque questo tRNA poteva leggere il codone di stop, vicino al gene per il tRNA (pyIT) c’era un gene simile a un’amminoacil- tRNA-sintetasi (pyIS) che però non era corrispondente esattamente a nessuna delle amminoacil- tRNA-sintetasi conosciute. Quindi sulla base di questa evidenza, la vostra ipotesi quale può essere? Com’è che la pirrolisina viene incorporata nella metilammina metiltransferasi quindi perché è proprio il 22esimo aa e non una modifica di un aa esistente? Il tRNA viene caricato con pirrolisina, e questo incorpora pirrolisina nella sintesi proteica è un aa proteinogenico anche se non universale. Noterete dall’immagine che vicino ai 2 geni, c’erano altri geni coinvolti nel processo: quale funzione potrebbero avere? Servono per la sintesi della pirrolisina, sono geni biosintetici per la pirrolisina. Questo è un esempio di come la vicinanza genica sia stata fondamentale per trovare questo particolare aa e la sua via biosintetica. La vicinanza genica è stata fondamentale anche per la scoperta del sistema CRISPR/Cas: osservando strani segmenti di cromosomi batterici in cui erano presenti ripetizioni palindromiche CRISPR e in cui c’erano dei geni associati (Cas=CRISPR associated) che assomigliavano a proteine note, ma senza essere esattamente corrispondenti. Assomigliavano a endonucleasi e tra i geni Cas vi è Cas (endonucleasi a guida a RNA) usatissima e importantissima a scopi biotecnologici.
Per concludere l’argomento dell’associazione sul cromosoma, vi faccio fare una distinzione che fanno anche questi programmi di predizione che riguarda l’orientazione relativa dei geni. Possiamo avere 3 casi: la stessa orientazione (tail to head), 2 geni divergenti (head to head), 2 geni convergenti (tail to tail). Dov’è più forte l’evidenza di associazione in questi 3 casi? Nella stessa orientazione (che sono tipicamente i geni in operone che possono essere co-trascritti); l’associazione tra i geni convergenti invece è quella meno forte; i geni divergenti possono indicare un’associazione perché se pensiamo alla trascrizione, essi possono avere un promotore in comune ed essere fattore trascrizionale per l’operone successivo. Cosa succede negli eucarioti? Ce l’abbiamo questa evidenza di vicinanza genica? Vi sono rari esempi di geni funzionalmente associati e vicini nel cromosoma, hanno quindi una somiglianza con l’operone batterico, lo ritroviamo solo in alcuni eucarioti come nei funghi e nel S.cerevisiae in cui ci sono 2 loci conosciuti in cui gli enzimi per una certa via metabolica sono messi vicini nel cromosoma (un es. è DAL). tutti i geni che codificano per la degradazione dell’allantoina sono messi vicini e ciò è successo durante l’evoluzione a partire da geni separati nel S.cerevisiae. Per quanto riguarda i mammiferi, l’unico caso che può indicarci un’associazione funzionale è questo delle coppie geniche head to head coppie in cui avete 2 geni trascritti in maniera divergente e in cui c’è un intervallo tra i 2 inizi della trascrizione (TSS) è meno di 1000bp, in questa regione abbiamo un promotore condiviso tra i 2 geni, si è visto che statisticamente hanno un’associazione funzionale e sono spesso trascritti nelle stesse condizioni (in H.sapiens circa un migliaio di head to head). Dal punto di vista numerico come posso associare un punteggio alla vicinanza di 2 geni diversi sul cromosoma? Prendo in considerazione la numerosità dei casi che osservo.
la semialdeide. Loro sapevano che c’era questa associazione biochimica che era suggerita dalla fusione genica nel batterio. Nell’uomo esiste l’idrossilisina che è un a.a che forma legami crociati nel collagene e ha una sua via degradativa. Loro hanno trovato i 2 prodotti di questi geni umani nella via degradativa cioè partendo dall’idrossilisina hanno osservato l’intermedio fosforilato e successivamente hanno ottenuto questa aldeide (che è anche l’intermedio della degradazione della lisina). Grazie alla cultura biochimica hanno colto il suggerimento bioinformatico. Per confermare l’ipotesi bioinformatica producono la proteina in forma isolata per poi testare l’attività della proteina in vitro, (questa però non è l’unica via). Un’altra possibilità oltre a studiare la proteina senza l’organismo (come i biochimici), è studiare l’organismo senza la proteina (come i genetisti). Infatti, dato che abbiamo 2 enzimi in una via metabolica potremmo fare il deleto del gene e si va a vedere il fenotipo, in questo caso l’accumulo dell’intermedio (non nell’uomo ma nell’organismo modello come ad es.il topo). Ultimo caso di evidenze: Co-occorrenza di geni differenti. Con co-occorrenza intendiamo la co- presenza o co-assenza di 2 geni A e B in una serie di organismi. Vado a vedere se nel caso in cui sia presente il gene A trovo anche il gene B o viceversa cioè se non c’è A non c’è neanche il gene B. Bisogna tener fisso l’ordine dei vari organismi, posso costruire dei vettori in cui 0 rappresenta l’assenza del gene o 1 la presenza del gene. Questo prende il nome di profilo filogenetico e serve per scoprire delle somiglianze, se ci sono probabilmente i 2 geni sono collegati (es. possono essere enzimi della stessa via metabolica, formano complesso, stessa via di trasduzione..). Es. sempre di una via metabolica: Questo sistema viene utile quando un certo processo non è universale ma avviene in alcuni organismi in maniera sparsa nell’evoluzione. Allora troviamo la co-occorrenza, questo è il caso della degradazione delle purine perché è una via metabolica che c’è in diversi organismi. Alcuni organismi eliminano le purine perché non hanno necessità di conservare l’azoto e l’uomo elimina le purine a livello di urato, i mammiferi fanno qualche passo in più arrivano
Evidenze di associazione funzionale:
3. Profili filogenetici (co-occorrenza)
Pellegrini, M et al. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. PNAS, 1999 Gene A Gene B 0 1 1 0 1 0 0 1 1 0 1 0 Degradazione ossidativa delle purine NH 3 CO 2 NH 3 CO 2 NH 3 CO 2 glycine X X X urate allantoin
all’allantoina e poi invece ci sono le piante e i funghi che riciclano l’azoto e quindi ottengono NH3, nei batteri ci sono situazioni varie in base alla nicchia ecologica. In questo caso si cercavano geni successivi all’urato ossidasi (UOX) il primo enzima che trasforma l’urato nel primo intermedio. Si sapeva che c’erano dei passaggi successivi per arrivare all’allantoina ma i geni erano sconosciuti. Se l’ossidasi è presente nella via metabolica ci saranno anche i 2 geni associati, co-presenti alla ossidasi. se la ossidasi è assente mancano anche il primo e il secondo gene. Da un confronto fatto anni fa emerge una somiglianza tra il profilo di Uox e Urah e Urad (che erano geni ipotetici, senza caratterizzazione, grazie a questa evidenza ora hanno una funzione) suggerendo che sono i geni successivi necessari per la via metabolica. Ciò è stato dimostrato con l’espressione diretta della proteina, Urah interviene nel primo passaggio, Urad nel secondo. Ancora una volta dimostrazione con un saggio in vitro della funzione dell’enzima. Le informazioni nelle banche dati crescono sempre di più e si trovano ultimamente associazioni interessanti come ad es. il trasportatore dello spermatozoo che esiste nell’uomo che si chiama CatSper.Ha una decina di subunità che compongono questo canale e tutti questi geni sono associati per co- occorrenza perchè in certi organismi come il mammifero è necessario per la fecondazione, mentre altri organismi (come gli uccelli, pesci) fanno a meno di questo trasportatore e quindi mancano di tutti i 10 geni per le subunità. Con gli enzimi comunque siamo più avvantaggiati nella validazione sperimentale dell’ipotesi, in questo modo è più semplice (il gruppo di ricerca del prof preferisce questo tipo di analisi) in altri casi diventa più difficile. Un legame evolutivo tra i geni della via metabolica urate O (^2) Uox urate O (^2) Uox Gene? Gene? Gene? Gene? pathway presente pathway assente urate ( S )- allantoin O 2 Complete enzymatic pathway in mouse Uox HIU hydrolase OHCU decarboxylase Urah Urad
- Mouse genes
- Recombinant expression in E. coli
- Biochemical evidence: (^13) C NMR, Circular Dichroism Ramazzina I, et al. Completing the uric acid degradation pathway through phylogenetic comparison of whole genomes. Nat Chem Biol. 2006 ( S )- allantoin