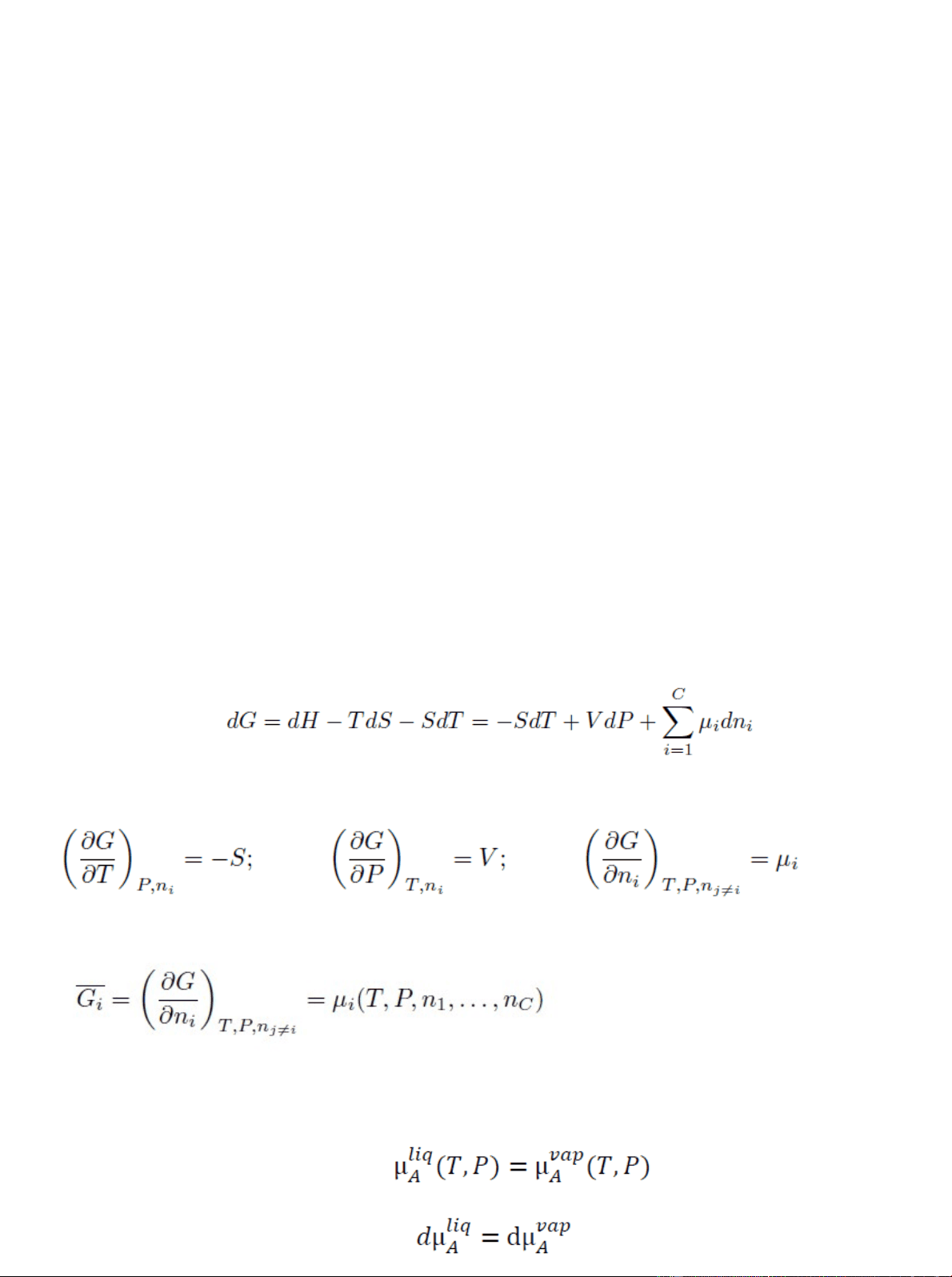Scarica Teoria Del Diritto E Dell'argomentazione e più Schemi e mappe concettuali in PDF di Scienze Sociali solo su Docsity!
Impianti chimici teoria
Operazioni unitarie:
Evaporazione Assorbimento/stripping Estrazione L/L Leaching (lisciviazione) L/S Cristallizzazione Adsorbimento Separazione ad opera di membrane Separazioni fisiche/meccaniche
Gradi di libertà del sistema:
GDL = Componenti − fasi + 2. I gradi di libertà indica in numero di variabili che possono agire indipendentemente
Termodinamica applicata a equilibri tra fasi fluide
Le operazioni unitarie operano trasformazioni o separazioni su sistemi non in equilibrio. Maggiore è lo scostamento dall’equilibrio, maggiore sarà la forza motrice del processo. Quantificare correttamente le condizioni di equilibrio per determinare il punto di arrivo della trasformazione e quantificarne la velocità, da cui poi dimensionare l’apparecchiatura. G = H − TS Da cui derivando: G soprassegnato viene definita energia libera di Gibbs parziale molare ed è pari al potenziale chimico della specie i nella fase data specificate le variabili da cui dipende. Condizioni di equilibrio:
Rappresentazione di variabili termodinamiche
Le proprietà volumetriche e calorimetriche dei sistemi omogenei a più componenti contengono come variabili aggiuntive, rispetto al caso dei sistemi a un componente, la n-pla ossia un vettore composizione di n componenti nella fase considerata (N1,N2,...,Nn). In generale, e al contrario di quanto accade per le miscele ideali, le proprietà volumetriche e calorimetriche di tali sistemi non sono esprimibili in modo adeguato conoscendo solo la composizione del sistema e le omonime proprietà dei componenti puri. Dal punto di vista pratico ciò si traduce in un volume di mescolamento ed entalpia di mescolamento diversi da zero. una generica proprietà (termica o volumetrica) di un sistema a più componenti si può esprimere correlando opportunamente:
- Le variabili di composizione del sistema
- Le omonime proprietà dei componenti puri
- Le omonime proprietà di tutti i sistemi binari che possono aver luogo Con questa ipotesi, il numero delle misure da effettuare per la valutazione, in un certo dominio delle variabili T,P,x , di una certa grandezza termica o volumetrica diminuisce drasticamente rispetto a quello prima visto: tali misure inoltre - e ciò costituisce un'altra non trascurabile semplificazione "strumentale" – riguardano solo sistemi a un componente e sistemi binari. Qmm è la proprietà di 1 solo, Qmn è la proprietà nel sistema binario pesate per le x
Correlazioni volumetriche per fasi gassose
Miscela ideale di gas ideali: vi è additività di V (ΔVmix =0) L’equazione di stato (EoS) espressa per il volume molare è: P v ¿ = RT ; d μ ¿ = v ¿ dP − s ¿ dT che^ a^ T^ cost^ : dT μ ¿ = v ¿ dP = RT P dP = RT dT lnP ( dP P loesprimo come dT lnP ) Miscela ideale di gas reali: vi è additività di V (ΔVmix =0) Si considera come l’ideale con un fattore di correzione EoS per gas reali
La relazione introdotta da Soave esprime il parametro attrattivo come prodotto tra il suo valore al punto critico e una funzione della temperatura ridotta tipica del composto considerato.
a = α ( T R )∗ ac [ T R = T / T c ]
L’espressione di α, inizialmente formulata da Soave nel 1972, introduce il fattore acentrico ω di Pitzer (distribuzione delle cariche interne alle molecole) come terzo parametro necessario per riprodurre correttamente le proprietà del composto, riconoscendo insufficienti le sole pressione e temperatura critiche. α ( T )=[ 1 + m ( 1 − T (^) R 0, )] 2 ;m =0,48+1,57 ω −0,176 ω 2 In seguito, numerosi autori hanno proposto altre espressioni di α, aumentando il numero dei parametri e complicando l’espressione originale di Soave per migliorare i risultati a temperature superiori a quella critica (estrapolazione di Boston – Mathias, 1980) e ottenere una riproduzione migliore della tensione di vapore dei composti polari. Miscela reale di gas reali Non vale l’additività dei volumi, quindi, occorre valutare le proprietà dei gas in miscela con parametri diversi dai composti puri. Si introdurrà non solo una deviazione dalla idealità in termini di fugacità del gas puro, ma anche per tener conto delle interazioni binarie. Si definirà quindi una fugacità della specie i in miscela, in generale diversa (a pari T e P) da quella del composto puro.
Fugacità
La definizione del potenziale chimico introdotta per i gas ideali può essere estesa ai gas reali sostituendo alla P un termine di fugacità. itegrando tra gli stati e uno stato di riferimento r supposto uguale per entrambi: μα − μr = RTln f α f r Pertanto, la definizione di condizione di equilibrio rispetto al trasferimento di massa tra due fasi (uguaglianza del potenziale chimico di i nelle due fasi) diventa:
E nel caso di ELV La fugacità di un gas o vapore si calcola scaricando la misura della deviazione dall’idealità su un coefficiente, detto coeff. di fugacità φi, tendente a 1 per P tendente a 0 (condizioni ideali) e valutabile mediante opportuni modelli. Nel caso di miscela ideale di gas reali φ si calcola come per il componente puro, nel caso di miscele non ideali dipenderà anche dalla composizione: mix ideale : φ dipende solo da P e T infatti si comporta come se fosse puro (yi=1) mix non ideale Miscele ideali di gas ideali Contributo negativo Miscele ideali o reali di gas reali
Dove Gi sono molari parziali Ricordando che: l’energia libera di Gibbs molare d’eccesso (g E ) rappresenta l’entità delle interazioni di miscelamento tra i componenti della miscela. Se è nulla i componenti non interagiscono e la miscela si comporta idealmente, se è >0 sono presenti forze repulsive che destabilizzano il sistema, se <0 sono presenti forze attrattive che lo portano verso una buca di potenziale. Una delle correlazioni più semplici per miscele binarie si basa su un solo parametro empirico A (Eq. Porter): sostituisco 1 in 2 e ottengo:
Equilibrio liquido vapore
Modelli per ELV La condizione di equilibrio L-V è espressa dalla condizione di uguaglianza delle fugacità. Per sistemi multicomponente si possono usare due approcci. I metodi del primo gruppo (metodi diretti o φ- φ) richiedono il calcolo del coefficiente di fugacità di entrambe le fasi.
I metodi del secondo gruppo (indiretti o γ-φ) utilizzano il coefficiente di fugacità per la fase vapore e il coefficiente di attività e la tensione di vapore per la fase liquida. Per il calcolo dei coefficienti di fugacità i metodi del primo gruppo utilizzano una EoS abbinata a regole di miscelazione in grado di descrivere, oltre alla fase vapore, anche la fase liquida, mentre i metodi del secondo gruppo utilizzano modelli per il calcolo dell’energia libera di eccesso. Confrontando i due approcci, i metodi del primo gruppo mostrano diversi vantaggi: l’approccio nei confronti delle diverse fasi è simmetrico quindi, qualunque sia la fase da descrivere, vengono calcolate le stesse grandezze, possono trattare qualunque tipo di sistema, incluse miscele contenenti composti supercritici, in un ampio campo di pressione e temperatura, comprese condizioni vicine al punto critico(impossibile per i metodi del secondo gruppo),Infine le equazioni di stato permettono di calcolare valori consistenti di importanti proprietà quali densità, entalpia, entropia e calore specifico. Caso 1: V ideale/ideale, L ideale. La fugacità del V si quantifica in termini di pressione parziale mediante l’equazione di Dalton. La fugacità del L si esprime mediantela legge di Raoult, che esprime la proporzionalità con la tensione di vapore. Caso 2: V ideale/ideale, L non ideale. La fugacità del L si quantifica correggendo la legge di Raoult con φ o γ.
Si definisce inoltre la volatilità relativa tra due composti: nel caso di idealità avrò che: quindi i composti sono separabili in funzione delle loro p°. nel caso di non idealità avrò che: portando alla formazione di azeotropi per certi valori di γ
Modelli per il calcolo dei coefficienti di fugacità
Vi = volume molare; V^ i volume molare parziale Volumi ideali questa relazione contempla tutte le casistiche di idealità e no Quindi utilizzando diverse EoS è possibile calcolare il coeff. di fugacità. Ad esempio facendo riferimento all’eq. del viriale: Bij coefficiente interazione binario Cijk coefficiente interazione ternaria O se si tronca l’eq. al primo termine (ok per P non troppo elevata):
Altri metodi definiscono i φi in funzione di ulteriori parametri, come ad esempio il fattore acentrico (Pitzer), definito come: Dove p° è la tensione di vapore alla T per cui TR = 0.7. Alternativamente sono disponibili metodi grafici per la valutazione di φ in funzione delle variabili ridotte. con ZR =^ Pc ∗ V (^) c T (^) c =0, se ZR è diverso da 0, Dove D è ricavato per via grafica
Modelli per il calcolo dei coefficienti di attività
- Simmetrica: condizione di idealità per tutti i componenti γi→1 per xi→1. Ok per due liquidi che rimangono tali in tutti il campo di composizioni.
- Asimmetrica: condizione di idealità per tutti i liquidi γi→1 per xi→1 , ma per il componente supercritico o soluto γi→1 per xi→0 (quando è puro non è più liquido). I modelli per il calcolo dei coefficienti di attività si dividono essenzialmente in due categorie: i modelli classici e i modelli a composizione locale. A questi si aggiungono i modelli a contributi di gruppo, i quali non si basano sulle interazioni binarie tra composti, ma tra i gruppi funzionali che li caratterizzano.
Modelli classici
Modello di Wohl Considera le interazioni binarie e ternarie (tra 3 molecole a due a due diverse: iij o ijj, ecc. La probabilità di interazione ijk è considerata ininfluente in miscele ternarie) tra le molecole costituenti la miscela, la cui entità è calcolata in funzione delle frazioni in volume φ a loro volta dipendenti dai volumi efficaci q (comprende anche la sfera di solvatazione). a: coefficienti di interazione (sommatoria dei volumi efficaci pesati per la frazione molare)
le forze repulsive uno ione hard tenderà a farsi circondare da molecole di acqua piuttosto che da altri ioni x 21 = frazione molare locale xi = frazione molare x (^) ji xii = x (^) j epx (− g (^) ji / RT ) xi epx (− gii / RT ) I parametri di interazione binari non vengono modificati in presenza di miscele a più di due componenti, in quanto vengono prese in considerazione solo le interazioni locali tra due specie. g è un termine energetico che quantifica l’interazione tra molecole uguali o diverse. Modello di Wilson Per tener conto della non casualità della distribuzione locale delle molecole Wilson suggerisce una relazione tra la frazione molare locale x 11 della molecola 1 e x 21 della molecola 2 nell’intorno della molecola 1 e le frazioni molari medie x 1 e x 2. Frazioni volumetriche totali Modello NRTL (non-random-two-liquids) Le equazioni NRTL sono ricavate basandosi sulla teoria dei due fluidi in cui si considerano due tipi di celle in una miscela binaria ciascuno centrato su una molecola diversa. L’energia libera residua delle celle contenenti la molecola di tipo 1 al centro è la somma pesata dalle frazioni molari locali delle energie di interazione di entrambe le molecole nei confronti della molecola 1.
I diversi modelli prevedono in modo simile il comportamento a diluizione infinita, ma deviano molto per medie composizioni. Adattando opportunamente αil modello NRTL riesce ad adattarsi meglio ai dati sperimentali ed è quindi più versatile specialmente per sistemi fortemente non ideali e che smiscelano in fase liquida. se mi trovo molto a dx o sx non ha senso utilizzare modelli complessi, userò modelli più semplici (i primi) che tanto non variano molto. 0,25<α<0,
I gruppi funzionali sono tabulati in gruppi e sottogruppi appartenenti ad una classe più ampia. Ogni sottogruppo ha parametri di volume e superficie, mentre le interazioni energetiche dei singoli gruppi sono ricondotte alla classe di appartenenza.
Regole di miscelazione
Utilizzando i metodi del primo gruppo basati sull’utilizzo di un’equazione di stato per descrivere sia la fase liquida che la fase vapore, è necessario fissare delle relazioni, dette regole di miscelazione, tra il parametro attrattivo e il covolume della miscela e dei composti puri. Le regole di miscelazione quadratiche classiche, con un solo parametro di interazione binaria kij, costante al variare di temperatura e composizione, introdotto nel calcolo del parametro attrattivo di miscela, permettono una descrizione accurata delle miscele di composti non polari o leggermente polari. In particolare, sono adatte alla descrizione di miscele regolari di idrocarburi e composti leggermente polari quali anidride carbonica e solfuro di idrogeno. La descrizione di sistemi contenenti sostanze polari risulta invece poco soddisfacente. Può essere significativamente migliorata dall’introduzione di un secondo parametro di interazione binaria dij per il calcolo del covolume della miscela, ma l’elevata correlazione tra i parametri binari determina valori assurdi degli stessi e del covolume, cioè della densità delle miscele liquide, nel caso di correlazione di dati sperimentali di miscele fortemente non ideali. L’estensione dei metodi del primo gruppo, per il calcolo degli equilibri di fase, a miscele contenenti sostanze polari è dovuta all’introduzione, da parte di Huron e Vidalnel 1979, di regole di miscelazione non quadratiche in cui i parametri della miscela sono legati all’energia libera di eccesso attraverso una relazione ricavabile dall’equazione di stato.
queste regole di miscelazione non vanno bene per i liquidi se non estrapolando dai casi a P infinita o 0
entalpia
La presenza di interazioni attrattive o repulsive tra i componenti della miscela comporta la variazione di entalpia della miscela stessa in seguito a miscelamento dei componenti (ovvero la non additività delle entalpie dei componenti puri). Pertanto, un’altra caratteristica delle miscele non ideali è ΔHmix ≠ 0.
Per un vapore/gas ideale: dove hi è del componente puro alla T di riferiemento E per una mix ideale l’entalpia è additiva: Per miscele non ideali l’entalpia di mescolamento viene calcolata come entalpia d’eccesso: Quindi utilizzando i vari modelli visti per il calcolo di γ si calcola h E : modello di Van Laar: Modello NRTL con parametri dipendenti da T:
Ricavare i parametri termodinamici di interesse
I problemi di equilibrio liquido vapore di miscele binarie, richiedono lo studio di alcune proprietà dei fluidi puri e della miscela.