Download Multiple Alignment Motif Finding - Lecture Notes | CMSC 423 and more Study notes Computer Science in PDF only on Docsity!
CMSC423: Bioinformatic Algorithms,
Databases and Tools
Lecture 13
multiple alignment
motif finding
Recap
• Multiple alignment is expensive – O(nk) for k
sequences of length n (use same DP as for pairwise
but on a k-dimensional matrix)
• Approximation algorithm (star alignment) can find a
solution in O(n
2
k
2
) which is at most twice worse than
the best alignment
Iterative alignment revisited
- Pick a sequence (e.g. SC) as a starting point
- Align S1 to it & build consensus for the alignment
- Take S2 and align it to the consensus (instead of SC)
- repeat...
- Problem: consensus (or any single sequence) ignores the other
sequences being aligned.
- Solution: keep track of % of each amino-acid aligned in each
column
- score of alignment to profile – combination of scores to each
AA.
Profile alignment
- Solution: keep track of % of each amino-acid aligned in each
column
- score of alignment to profile – combination of scores to each
AA.
- Score(prof1, prof2) = weighted average of all pairs of amino-
acids
S1 YFPHF-DLS-----HGSAQVKAHGKKVG-----DALTLAVAHLDDLPGAL S2 YFPHF-DLS-----HG-AQVKG—GKKVA-----DALTNAVAHVDDMPNAL S3 FFPKFKGLTTADQLKKSADVRWHAERII-----NAVNDAVASMDDTEKMS S4 LFSFLKGTSEVP--QNNPELQAHAGKVFKLVYEAAIQLQVTGVVVTDATL
50% S 25% N 100% F 25% -
75% A 25% Q
Iterative alignment
• Take sequences si in order:
- align s1 with sc - results in gaps being inserted in both sequences
- align s2 with sc - if gaps must be inserted – insert in previously aligned sequences
- and so on (note: if gaps coincide with previously introduced gaps no need to change previously aligned sequences)
SC YFPHFDLSHGSAQVKAHGKKVGDALTLAVGHLDDLPGAL
SC YFPHFDLSHGSAQVKAHGKKVGDALTLAVGHLDDLPGAL S1 YFPHFDLSHG-AQVKG--KKVADALTNAVAHVDDMPNAL
SC YFPHF-DLS-----HGSAQVKAHGKKVG-----DALTLAVAHLDDLPGAL S1 YFPHF-DLS-----HG-AQVKG—GKKVA-----DALTNAVAHVDDMPNAL S2 FFPKFKGLTTADQLKKSADVRWHAERII-----NAVNDAVASMDDTEKMS
SC YFPHF-DLS-----HGSAQVKAHGKKVG-----DALTLAVAHLDDLPGAL S1 YFPHF-DLS-----HG-AQVKG—GKKVA-----DALTNAVAHVDDMPNAL S2 FFPKFKGLTTADQLKKSADVRWHAERII-----NAVNDAVASMDDTEKMS S3 LFSFLKGTSEVP--QNNPELQAHAGKVFKLVYEAAIQLQVTGVVVTDATL
CLUSTALW
• Compute pairwise distances between strings
• Build phylogenetic tree
• Build iterative alignment by following tree edges
s
s
s s
s
s
Biological relevance of multiple alignments
Motif finding
Motif finding...example
From genetics.mgh.harvard.edu/sheenlab/
TTAGAGGTTGACTATTCAACTTTTGAGGAGGCCTAG TAGAGC
AGCCGACTTGCAACTTAGGCGTGGTCAGTGCCCTAA TAGAGC
GGCCTATTTGGGCCACTTAGACCTTCAACTTTTGCA TAGAGC
CCACAGTTAGATGTCCAAAAGACAAATATAGAGGGC TAGAGC
ACACGGACTGCGTTCAATGCTTACAGCAGATTGAGT TAGAGC
TTCAAAGACTTGACTATTGTTCAACTTTGAAGACTA TAGAGC
Promoter region Gene
Motif “sequence logo”
Finding motifs – Gibbs sampling
• Observations:
- since no gaps – all motifs have equal length (assume known value - m)
- exhaustive search of promoter region is impractical: all combinations of substrings of length m among k sequences of length L = (L – m + 1)k
- Solution: random search
- Pick random substring of length m from each of the strings
- Construct multiple alignment (easy since no gaps) and compute
profile
- Pick random sequence s and remove from multiple alignment.
Recompute profile.
- Within removed sequence, search for best fit to profile and
insert into alignment
- Repeat until profile does not improve
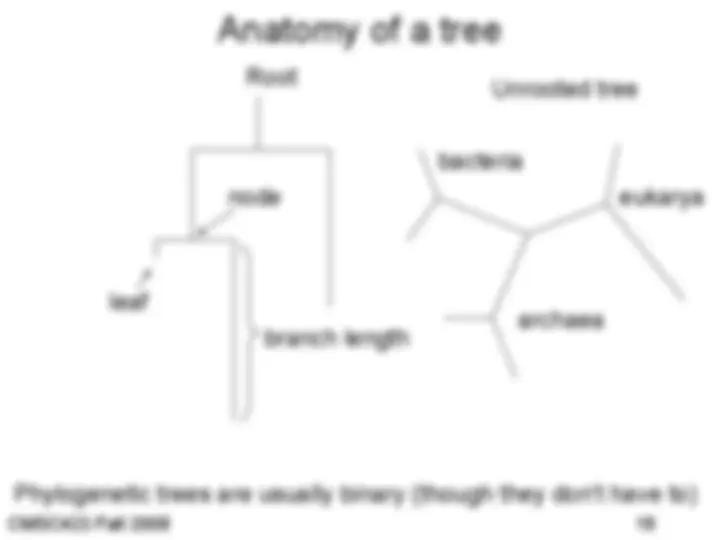
Phylogenetic trees
Phylogenetic trees – how evolution works
• http://www.tolweb.org/tree/ - the tree of life
Phylogeny questions
- Given several organisms & a set of features (usually sequence,
but also morphological: wing shape/color...)
- A. Given a phylogenetic tree – figure out what the ancestors
looked like (what are the features of internal nodes)
- B. Find the phylogenetic tree that best describes the common
evolutionary heritage of the organisms
wings, feathers, teeth claws, no wings, fur
A C
B A
B
B
A
C C