Partial preview of the text
Download Notes on Motif Finding Microarray Data Analysis | CMSC 423 and more Study notes Computer Science in PDF only on Docsity!
CMSC423: Bioinformatic Algorithms,
Databases and Tools
Lecture 20
Motif finding
Microarray data analysis
forward-backward why backward
N - # of genes, L - # length of upstream region, K-motif length(L-K+1)
N
possible choices
Probabilistic search
Outline:
– Pick a set of random k-mers
(one from each sequence)
– Build a multiple-alignment
profile – frequency of eachnucleotide at each of the kpositions
– Remove one sequence at
random and find the k-merwithin it that best matches theprofile (p(k-mer|profile)=product of frequencies for k-mer nucleotides in profile table)
– Recompute profile and repeat
T
G
C
A
Types of microarrays
By technology
– Spotted– Affymetrix– Nimblegen– Illumina
By information
– cDNA (genes or parts of genes)– DNA (e.g. sequencing by hybridization)– Tiling arrays (whole genome)– Protein
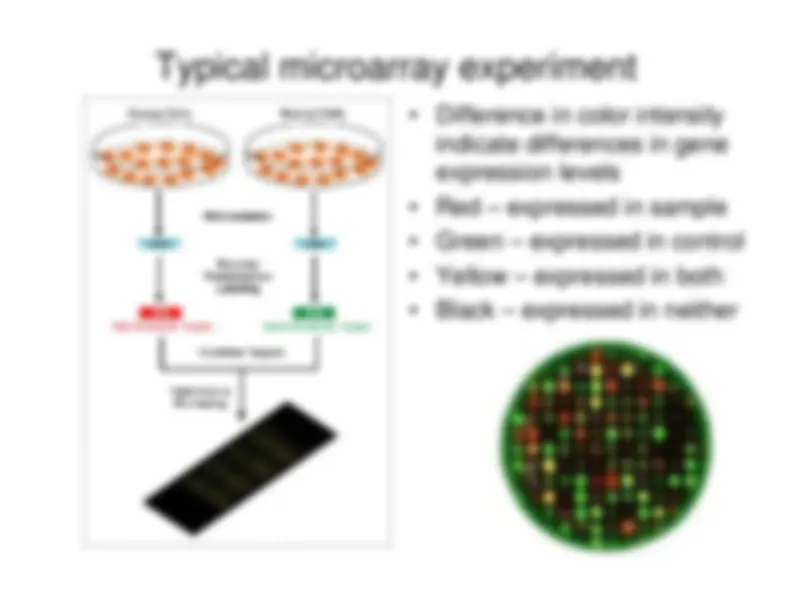
Typical microarray experiment
Difference in color intensityindicate differences in geneexpression levels
Red – expressed in sample
Green – expressed in control
Yellow – expressed in both
Black – expressed in neither
Data clustering
Agglomerative
– Start with single observations– Group similar observations into the same cluster
Divisive
– All datapoints start in the same cluster– Iteratively divide cluster until you find good clustering
Hierarchical
– Build a tree – leaves are datapoints, internal nodes
represent clusters
Measures of goodness of clustering
Homogeneity
– All points in a cluster must be similar
Separation
– Points in different clusters are disimilar
Hierarchical clustering
UPGMA (remember from phylogenetic trees?)
– compute distance between genes (e.g. euclidean distance of
expression vectors)
– join most similar genes– repeat– Key element – compute distance between a gene and a
cluster, or between two clusters – average distance betweenall genes in the two clusters