¡Descarga Dosis y administración del medio de contraste en angiografía y más Transcripciones en PDF de Marketing en Farmacia solo en Docsity!
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Ultravist 300 mg/ml solución inyectable y para perfusión en vial
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
1 ml de solución inyectable contiene 623 mg de iopromida (equivalente a 300 mg de iodo).
Excipientes con efecto conocido: 1 ml de solución inyectable contiene 0,2549 mg (equivalente a 0,01109 mmol) de sodio. Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable y para perfusión en vial. Solución clara, de incolora a amarillo pálido.
Las características físico-químicas de la solución inyectable de Ultravist, a la concentración de 300 mg I/ml se indican en la tabla a continuación.
Osmolalidad (osm/kg H 2 O) a 37 C 0, Viscosidad (mPa.s) a 20C a 37C
Densidad (g/ml) a 20ºC a 37ºC
pH 6,5-8,
4. DATOS CLÍNICOS
4.1. Indicaciones terapéuticas
Este medicamento es únicamente para uso diagnóstico
Ultravist 300 mg/ml está indicado en adultos para:
realce del contraste en tomografía computarizada (TC) arteriografía convencional flebografía convencional de extremidades angiografía por sustracción digital (ASD)
- urografía intravenosa
- mamografía con contraste en mujeres adultas para evaluar y detectar lesiones de mama conocidas o sospechadas, como complemento de la mamografía (con o sin ecografía) o como alternativa a la resonancia magnética (RM) cuando la RM está contraindicada o no está disponible. artrografía e histerosalpingografía
Ultravist 300 mg/ml está indicado en población pediátrica de 0-18 años para:
- realce de contraste en tomografía computarizada (TC)
- angiografía por sustracción digital (ASD)
- urografía intravenosa
- arteriografía convencional
- flebografía
4.2. Posología y forma de administración
Posología
Las dosis indicadas a continuación constituyen únicamente recomendaciones y representan las dosis habituales para un adulto de 70 kg de peso. Las dosis se expresan para inyección única o por kilo de peso corporal (kg p.c.).
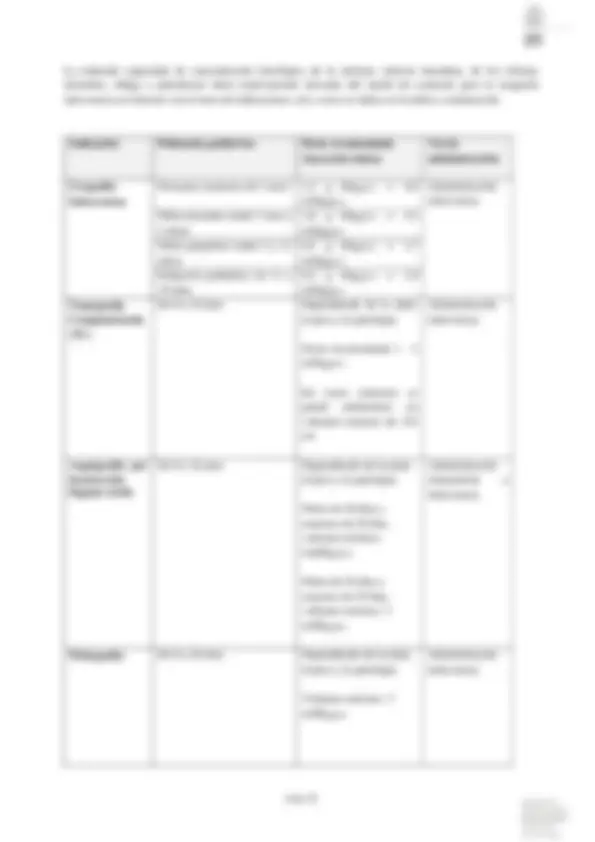
Indicación Dosis Recomendada (inyección única)
Dosis Máxima Total
Arteriografía Convencional cayado aórtico 50 – 80 ml 1,5 g I por kg.p.c. vascular selectiva 6 – 15 ml 1,5 g I por kg.p.c. carotidea retrógrada
30 – 40 ml 1,5 g I por kg.p.c.
Aortografía Convencional
Administración por vía intraarterial o intravenosa.
torácica 50 –^ 80 ml^ 1,5 g I por kg.p.c. abdominal 40 –^ 60 ml^ 1,5 g I por kg.p.c. Arteriografía de Extremidades superiores 6 –^ 12 ml^ 1,5^ g I por kg.p.c. inferiores 20 –^ 30 ml^ 1,5 g I por kg.p.c. Flebografía de Extremidades superiores 14 –^ 30 ml^ 1,5 g I por kg.p.c. inferiores 30 –^ 60 ml^ 1,5 g I por kg.p.c. Angiografía por Sustracción Digital (ASD) intravenosa 30 –^ 60 ml^ 1,5 g I^ por kg.p.c. Velocidad del flujo: 8 - 12 ml/seg en la vena cubital; 10- 20 ml/seg por catéter en la vena cava únicamente para la visualización de los grandes vasos del tronco. Puede reducirse la cantidad de medio de contraste presente en las venas y ser a la vez diagnóstica, administrando una solución isotónica de cloruro sódico en bolo inmediatamente después. intraarterial 2 – 25 ml 1,5 g I por kg.p.c. En la ASD intraarterial son suficientes volúmenes más pequeños y concentraciones más bajas de iodo que en la técnica intravenosa. Cuanto más selectiva sea la angiografía, menores dosis de medio de contraste se necesitarán. Por ello, este método se recomienda en pacientes con una función renal restringida. Tomografía Computarizada (TC)
La reducida capacidad de concentración fisiológica de la nefrona, todavía inmadura, de los riñones infantiles, obliga a administrar dosis relativamente elevadas del medio de contraste para la urografía intravenosa en relación con el resto de indicaciones, tal y como se indica en la tabla a continuación.
Indicación Población pediátrica Dosis recomendada (inyección única)
Vía de administración
Urografía Intravenosa
Neonatos (menores de 1 mes) 1,2 g I/kg.p.c. = 4, ml/kg.p.c.
Administración intravenosa Niños lactantes (entre 1 mes y 2 años)
1,0 g I/kg.p.c. = 3, ml/kg.p.c Niños pequeños (entre 2 y 11 años)
0,5 g I/kg.p.c. = 1, ml/kg.p.c Población pediátrica de 11 a 18 años
0,3 g I/kg.p.c. = 1, ml/kg.p.c Tomografía Computarizada (TC)
De 0 a 18 años Dependiendo de la edad, el peso y la patología.
Dosis recomendada 1 - 3 ml/kg.p.c.
En casos extremos se puede administrar un volumen máximo de 125 ml.
Administración intravenosa
Angiografía por Sustracción Digital (ASD)
De 0 a 18 años Dependiendo de la edad, el peso y la patología.
Niños de 28 días y menores de 28 días, volumen máximo: 4ml/Kg p.c.
Niños de 29 días y mayores de 29 días, volumen máximo: 5 ml/Kg p.c.
Administración intraarterial e intravenosa
Flebografía De 0 a 18 años^ Dependiendo de la edad, el peso y la patología.
Volumen máximo: 3 ml/Kg p.c.
Administración intravenosa
Arteriografía Convencional
De 0 a 18 años Dependiendo de la edad, el peso y la patología.
Niños de 28 días y menores de 28 días, volumen máximo: 4 ml/Kg p.c.
Niños de 29 días y mayores de 29 días, volumen máximo: 5 ml/Kg p.c.
Administración intraarterial
Recién nacidos (<1 mes) y niños lactantes (de 1 mes a 2 años): Los niños menores de 1 año de edad, y especialmente los recién nacidos, son susceptibles de padecer trastornos electrolíticos y alteraciones hemodinámicas. Se deberá prestar especial atención en lo relacionado con la dosis del medio de contraste a administrar, con la técnica del procedimiento radiológico y con el estado del paciente.
Las dosis recomendadas en neonatos, niños lactantes, niños pequeños y población pediátrica de 11 a 18 años, no deben sobrepasarse.
Pacientes con insuficiencia hepática: No es necesario realizar un ajuste de la dosis. Véase advertencias y precauciones en sección 4.4.
Pacientes con insuficiencia renal: Debido a que la iopromida es excretada casi exclusivamente de forma inalterada por los riñones, la eliminación de iopromida se encuentra prolongada en pacientes con insuficiencia renal. A fin de reducir el riesgo de lesión renal adicional inducida por el medio de contraste, en pacientes con insuficiencia renal pre- existente, se debe emplear la menor dosis diagnóstica (ver también sección 4.4).
Forma de administración
Información general:
Recomendaciones dietéticas Se puede mantener una dieta normal hasta dos horas antes de la administración del medio de contraste. Durante las 2 horas previas a la administración, el paciente debe abstenerse de comer para reducir el riesgo de aspiración ya que las náuseas y vómitos son posibles reacciones adversas conocidas de estos medios de contraste.
Calentamiento del medio de contraste previo a su administración Los medios de contraste que se calientan a la temperatura corporal antes de su administración se toleran mejor y pueden administrarse más fácilmente debido a la disminución de su viscosidad. Para más información ver sección 6.6.
Administración por vía intraarterial e intravenosa:
Si se siguen las directrices posológicas anteriores y se inyecta Ultravist 300 durante 1 - 2 minutos, normalmente el parénquima renal se opacifica de forma intensa tras 3-5 minutos y la pelvis renal con el tracto urinario en 8 a 15 minutos, tras el comienzo de la administración. Dentro de estos intervalos, se debe elegir el tiempo más próximo a la inyección para los pacientes más jóvenes y el tiempo más alejado para los de mayor edad.
En recién nacidos, lactantes y niños pequeños, se recomienda realizar la primera radiografía, a a los 2 - 3 minutos después de la administración del medio de contraste.
Artrografía e histerosalpingografía:
Este medicamento debe administrarse exclusivamente por vía intraarticular o intrauterina.
La administración intraarticular o intrauterina de este medicamento debe realizarse exclusivamente por personal autorizado. La prueba debe realizarse bajo supervisión médica.
Durante la artrografía y la histerosalpingografía se recomienda realizar las inyecciones del medio de contraste bajo control fluoroscópico.
4.3. Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6. (incluida alergia al iodo). Hipertiroidismo clínico. No deben realizarse histerosalpingografías durante el embarazo, como en todos los procedimientos radiológicos, o en presencia de procesos inflamatorios agudos en la cavidad pélvica.
4.4. Advertencias y precauciones especiales de empleo
Para todas las indicaciones
Reacciones de Hipersensibilidad Ultravist puede estar relacionado con reacciones anafilactoides/ de hipersensibilidad y otras reacciones idiosincráticas que se manifiestan como síntomas cardiovasculares, respiratorios o cutáneos.
Las reacciones similares a las alérgicas van desde un rango de leves a graves, incluyendo la aparición de shock (ver sección 4.8). La mayoría de estas reacciones ocurren en la primera media hora, tras la administración. Sin embargo, pueden aparecer reacciones retardadas (tras horas o días). El riesgo de reacciones de hipersensibilidad es mayor en caso de:
- reacciones previas a medios de contraste
- historia de asma bronquial u otros trastornos alérgicos
Se debe valorar cuidadosamente el balance riesgo/beneficio, particularmente de los pacientes con hipersensibilidad conocida a Ultravist o a alguno de sus excipientes, o con una historia de reacción de hipersensibilidad previa a cualquier otro medio de contraste iodado debido a un incremento del riesgo de aparición de reacciones de hipersensibilidad (incluyendo reacciones graves).
Sin embargo, dichas reacciones son variables e impredecibles.
Los pacientes que experimentan éstas reacciones mientras toman beta bloqueantes pueden ser resistentes al tratamiento con beta agonistas (ver sección 4.5).
En caso de aparición de una reacción de hipersensibilidad grave, los pacientes con enfermedades cardiovasculares son más susceptibles a desenlaces graves o fatales. Debido a la posibilidad de aparición de reacciones de hipersensibilidad graves después de la administración, se recomienda la observación de los pacientes una vez finalizado el procedimiento diagnóstico. Es necesario que se dispongan de las medidas de urgencia adecuadas para todos los pacientes.
Si se administra premedicación en estos pacientes (con predisposición alérgica, con asma bronquial o con antecedentes de alergia a otros medios de contraste), se recomienda la utilización de un régimen con corticosteroides.
Reacciones adversas cutáneas graves (RACG) Se han notificado, con una frecuencia no conocida y asociadas a la administración de iopromida, reacciones adversas cutáneas graves (RACG) tales como síndrome de Stevens-Johnson (SSJ), necrólisis epidérmica tóxica (NET), reacción a fármaco con eosinofilia y síntomas sistémicos (DRESS) y pustulosis exantemática generalizada aguda (AGEP), que pueden ser potencialmente mortales o mortales.
Se debe informar a los pacientes sobre los signos y síntomas y se debe vigilar estrechamente la aparición de reacciones cutáneas.
En niños, la presentación inicial de una erupción puede confundirse con una infección, y los médicos deben tener en cuenta la posibilidad de una reacción a iopromida en niños que desarrollen signos de erupción y fiebre.
La mayoría de estas reacciones se produjeron en un plazo de 8 semanas (AGEP, 1-12 días; DRESS, 2- semanas; SSJ/NET, entre 5 días y 8 semanas).
Si el paciente ha sufrido una reacción grave tal como SSJ, NET, AGEP o DRESS con el uso de iopromida, no se debe volver a administrar iopromida a este paciente en ningún momento.
Disfunción tiroidea
Se debe valorar cuidadosamente el balance riesgo/beneficio, particularmente en pacientes con diagnóstico o sospecha de hipertiroidismo o bocio, debido a que los medios de contraste iodados pueden inducir hipertiroidismo y crisis tirotóxicas. Si se planea administrar un medio de contraste iodado en estos grupos de pacientes de riesgo, se debería valorar la función tiroidea antes de la administración de Ultravist, y/o la medicación tirostática preventiva.
Se han notificado pruebas de función tiroidea indicativas de hipotiroidismo o supresión tiroidea transitoria tras la administración de contraste iodado en pacientes adultos y pediátricos. Debe evaluarse el riesgo potencial de hipotiroidismo en pacientes con enfermedades tiroideas conocidas o sospechadas antes de utilizar medios de contraste iodados.
Se recomienda monitorizar la función tiroidea en neonatos, especialmente en prematuros, que han sido expuestos a Ultravist a través de la madre durante el embarazo o en el periodo neonatal, debido a que una exposición a un exceso de iodo puede causar hipotiroidismo, que posiblemente requiera tratamiento.
Trastornos del SNC Los pacientes con trastornos del SNC pueden tener un mayor riesgo de padecer complicaciones neurológicas relacionadas con la administración de iopromida. Las complicaciones neurológicas son más frecuentes con la realización de angiografía cerebral y procedimientos relacionados. Se ha notificado la aparición de encefalopatía con el uso de iopromida (ver sección 4.8). La encefalopatía por contraste se puede manifestar por síntomas y signos de disfunción neurológica tales como cefalea,
Administración por vía intraarterial o intravenosa
Lesión renal aguda La lesión renal aguda post-contraste (LRA-PC), se presenta como una insuficiencia transitoria de la función renal, y puede ocurrir después de la administración por vía intraarterial o intravenosa de Ultravist. En algunos casos, puede aparecer una insuficiencia renal aguda.
Los factores de riesgo incluyen, p. ej.: insuficiencia renal preexistente (ver sección 4.2, subsección Pacientes con insuficiencia renal ) deshidratación (ver sección 4.4, subsección Hidratación ) diabetes mellitus mieloma múltiple / paraproteinemia dosis altas y/o repetidas de Ultravist.
Los pacientes con insuficiencia renal moderada a grave (TFG estimada 44-30 mL/min/1,73 m^2 ) o grave (TFG estimada <30 mL/min/1,73 m^2 ) tienen mayor riesgo de sufrir una lesión renal aguda post-contraste (LRA-PC) con la administración de contraste intraarterial y la exposición renal de primer paso. Los pacientes con insuficiencia renal grave (TFG estimada <30 mL/min/1.73 m^2 ) tienen mayor riesgo de LRA- PC con la administración de contraste intravenoso o intraarterial con la exposición renal de segundo paso (ver sección 4.4, subsección Hidratación ). Se puede administrar Ultravist para exploraciones radiológicas a los pacientes en diálisis, sin función renal residual, ya que los medios de contraste iodados se eliminan en el proceso de diálisis.
La exploración con medios de contraste se decidirá con un criterio muy riguroso en casos de alteraciones graves de las funciones hepática o renal (manteniendo al paciente debidamente hidratado).
Sólo se administrará si es absolutamente necesario en pacientes con alteraciones graves de la función renal incluyendo pacientes con enfermedad hepática grave con síndrome hepatorrenal y a aquellos que van a ser sometidos a trasplante hepático, ya que pueden tener un retraso significativo en el aclaramiento del medio de contraste.
En pacientes con insuficiencia renal terminal, los medios de contraste no iónicos pueden eliminarse del organismo mediante hemodiálisis.
Diabetes mellitus La administración de medios de contraste iodados en pacientes diabéticos con daño renal preexistente predispone a disfunción renal. Puede aparecer acidosis láctica en pacientes en tratamiento con biguanidas (ver sección 4.5).
Patología cardiovascular Hay un mayor riesgo de que se produzcan cambios hemodinámicos clínicamente relevantes y arritmias en pacientes con patología cardiaca significativa o enfermedad coronaria severa.
La inyección intraarterial o intravenosa de Ultravist puede precipitar la aparición de un edema pulmonar en pacientes con insuficiencia cardiaca.
Feocromocitoma Los pacientes con feocromocitoma tienen riesgo de desarrollar una crisis hipertensiva.
Pacientes con trastornos autoinmunes
Se ha informado de casos de vasculitis graves o de un síndrome de tipo Stevens-Johnson, en pacientes con trastornos autoinmunes previos.
Miastenia gravis La administración de Ultravist puede agravar los síntomas de la miastenia gravis.
Fenómenos tromboembólicos Una propiedad de los medios de contraste radiológicos no iónicos es su escasa interferencia sobre las funciones fisiológicas normales. Como consecuencia de ello, su actividad anticoagulante in vitro es menor que la de los medios de contraste iónicos. Numerosos factores, además del medio de contraste, tales como la duración del procedimiento a realizar, el número de inyecciones, el tipo de material del catéter y de la jeringa, la patología subyacente del paciente y la medicación concomitante, pueden contribuir al desarrollo de acontecimientos tromboembólicos. Por consiguiente, todo ello debe ser tenido en cuenta cuando se lleve a cabo un procedimiento de cateterismo vascular, debiéndose prestar especial atención a la técnica angiográfica empleada e irrigar frecuentemente el catéter con suero salino fisiológico (añadiendo heparina, siempre que sea posible), así como minimizar la duración del procedimiento, con el objeto de minimizar el riesgo de acontecimientos tromboembólicos relacionados con el procedimiento diagnóstico realizado.
Se ha informado de que la utilización de jeringas de plástico en lugar de jeringas de cristal disminuye, pero no elimina, la posibilidad de que se produzcan fenómenos de coagulación in vitro.
Se aconseja tener precaución en los pacientes con homocistinuria debido al riesgo de inducir acontecimientos tromboembólicos.
Mieloma múltiple o paraproteinemia de Waldestrom Los pacientes con mieloma múltiple o paraproteinemia de Waldestrom tienen una mayor predisposición a presentar una insuficiencia transitoria de la función renal tras la administración por vía intraarterial o intravenosa del medio de contraste. En raras ocasiones puede aparecer una insuficiencia renal aguda (ver insuficiencia renal).
Mamografía con contraste (CEM) La mamografía con contraste conlleva una mayor exposición de la paciente a la radiación ionizante que la mamografía estándar. La dosis de radiación depende del grosor de la mama, del tipo de dispositivo mamográfico y de los ajustes del sistema del dispositivo. La dosis global de radiación de la CEM se mantiene por debajo del umbral definido por las directrices internacionales para la mamografía (por debajo de 3 mGy).
Artrografía e histerosalpingografía
Es necesario excluir toda posibilidad de embarazo antes de realizar una histerosalpingografía.
La inflamación de las trompas uterinas puede aumentar el riesgo de reacciones tras la histerosalpingografía.
4.5. Interacción con otros medicamentos y otras formas de interacción
Biguanidas (metformina): En pacientes con insuficiencia renal aguda o insuficiencia crónica grave, la eliminación de biguanidas puede verse disminuida, dando lugar a la acumulación y desarrollo de acidosis láctica. Como el uso de Ultravist puede dar lugar a una insuficiencia renal o un agravamiento de la misma, los pacientes tratados con metformina pueden tener un riesgo más elevado de desarrollar acidosis láctica,
Debe valorarse la relación riesgo-beneficio antes de administrar un contraste iodado teniendo en cuenta la sensibilidad del tiroides fetal por el iodo, ya que la sobrecarga aguda de iodo tras la administración de un contraste iodado a la madre puede provocar disfunción tiroidea fetal.
Lactancia:
No se ha investigado la seguridad de Ultravist en lactantes. La excreción de los medios de contraste en la leche humana es escasa. No es previsible daño alguno para el lactante (ver también sección 4.4 - Disfunción tiroidea ).
4.7. Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas.
4.8. Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad global de Ultravist está basado en los datos obtenidos de estudios previos a la comercialización, en más de 3.900 pacientes y de estudios postautorización en más de 74.000 pacientes, así como de datos de notificación espontánea y de la bibliografía.
Las reacciones adversas más frecuentemente observadas (> 4 %) en pacientes recibiendo Ultravist son cefalea, náuseas y vasodilatación.
Las reacciones adversas más graves observadas en pacientes recibiendo Ultravist son shock anafiláctico, parada respiratoria, broncoespasmo, edema laríngeo, edema faríngeo, asma, coma, infarto cerebral, accidente cerebrovascular, edema cerebral, convulsión, arritmias, parada cardiaca, isquemia de miocardio, infarto de miocardio, insuficiencia cardiaca, bradicardia, cianosis, hipotensión, shock, disnea, edema pulmonar, insuficiencia respiratoria y aspiración.
Lista tabulada de reacciones adversas
Las reacciones adversas observadas con Ultravist están representadas en la tabla a continuación. Están clasificadas de acuerdo con la clasificación de órgano-sistema. Para describir una determinada reacción, sus sinónimos y trastornos relacionados se utiliza el término MedDRA más apropiado.
Las reacciones adversas obtenidas de ensayos clínicos se clasifican de acuerdo a sus frecuencias. Los grupos de frecuencias están definidos de acuerdo con el siguiente convenio:
muy frecuentes: ≥1/
frecuentes: ≥ 1/100 a < 1/
poco frecuentes: ≥ 1/1.000 a < 1/
raras: ≥ 1/10.000 a < 1/1.
muy raras: < 1/10.
Las reacciones adversas identificadas únicamente durante el seguimiento post-comercialización, y para las cuales no se ha podido estimar ninguna frecuencia, se listan bajo “frecuencia no conocida”.
Tabla 1: Reacciones adversas notificadas en ensayos clínicos o durante el seguimiento post- comercialización en pacientes tratados con Ultravist
Clasificación de órganos del sistema
Muy frecuentes Frecuentes^ Poco frecuentes^ Raras^
Muy Raras
Frecuencia no conocida Trastornos del sistema inmunológico
Reacciones de hipersensibilidad / anafilactoides (shock anafiláctico§, parada respiratoria§, broncoespasmo, edema laríngeo o faríngeo* o facial o lingual§, espasmo§ laríngeo o faríngeo, asma§*, conjuntivitis§, lagrimeo§, estornudos, tos, edema de mucosas, rinitis§, ronquera§, irritación de garganta§, urticaria, prurito, angioedema) Trastornos endocrinos Crisis tirotóxica, Trastorno tiroideo Trastornos psiquiátricos Ansiedad, Trastornos del sistema nervioso
Mareos, Cefalea, Disgeusia
Reacción vasovagal, Estado confusional, Nerviosismo, Parestesia / hipoestesia, Somnolencia
Coma, Isquemia o infarto cerebral, Accidente cerebrovascu lar, Edema cerebrala, Convulsión*, Ceguera cortical transitoriaa, Pérdida de la consciencia, Agitación, Amnesia,
Clasificación de órganos del sistema
Muy frecuentes Frecuentes^ Poco frecuentes^ Raras^
Muy Raras
Frecuencia no conocida Trastornos musculoesqueléticos y del tejido conjuntivo
Síndrome compartimen tal en caso de extravasación a
Trastornos renales y urinarios
Insuficiencia renala Fallo renal agudoa Trastornos generales y alteraciones en el lugar de administración
Dolor, Reacciones en el lugar de inyección (diferentes tipos, p. ej. dolor, sensación de calor§, edema§, inflamación§ y lesión§^ del tejido blando en caso de extravasación ), Sensación de calor
Edema Malestar general, Escalofríos, Palidez,
Exploraciones complementarias
Fluctuacione s de la temperatura corporal
***** Se han notificado casos mortales o que han puesto en peligro la vida.
a (^) Sólo con la administración por vía intraarterial o intravenosa
§ (^) Reacciones identificadas sólo durante el seguimiento post-comercialización (frecuencia no conocida)
La mayoría de las reacciones después del uso del medio de contraste en cavidades corporales aparecen algunas horas después de la administración.
Efectos de clase La anestesia general está indicada para realizar la exploración en algunos pacientes seleccionados; sin embargo, se ha descrito una alta incidencia de reacciones adversas en dichos pacientes, que se ha atribuido a la incapacidad del paciente para distinguir entre reacciones adversas propiamente dichas y efectos hipotensivos de la anestesia que prolonga el tiempo de circulación e incrementa la duración de la exposición al medio de contraste.
Notificación de sospechas de reacciones adversas Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de medicamentos de Uso Humano: https://notificaram.es.
4.9. Sobredosis
Los resultados de los estudios de toxicidad aguda en animales no indican riesgo alguno de intoxicación aguda tras la administración de Ultravist.
Sobredosis por vía intraarterial o intravenosa:
Los síntomas pueden incluir alteraciones del equilibrio hidroelectrolítico, fallo renal, complicaciones cardiovasculares y pulmonares.
En caso de sobredosis intraarterial o intravenosa accidental se recomienda monitorizar la función renal y el balance hidroelectrolítico. El tratamiento de la sobredosis debe ir dirigido hacia el soporte de las funciones vitales.
Ultravist es dializable.
En caso de una sobredosis intraarterial o intravenosa accidental en humanos, la pérdida de agua y electrolitos debe compensarse mediante infusión. La función renal debe vigilarse durante al menos los 3 días siguientes a la realización de la prueba. Si es necesario, se puede utilizar la hemodiálisis para eliminar del organismo del paciente la mayor parte del medio de contraste.
5. PROPIEDADES FARMACOLÓGICAS
5.1. Propiedades farmacodinámicas
Grupo farmacoterapéutico: medios de contraste para rayos-X de baja osmolaridad, hidrosolubles y nefrotrópicos. Código ATC: V08AB05.
El compuesto que proporciona el contraste en las formulaciones de Ultravist es la iopromida, un derivado del ácido isoftálico triiodado en el que el iodo firmemente unido absorbe los rayos-X. La iopromida es un medio de contraste radiológico triiodado, no iónico, hidrosoluble, con un peso molecular de 791,12.
Mamografía con contraste (CEM)
Nueve estudios, que incluían a 1.531 pacientes, se centraron en el rendimiento diagnóstico en entornos clínicos relevantes.
En estudios que evaluaron lesiones sospechosas, la CEM mostró una sensibilidad del 96,9% al 100% y una especificidad del 69,7% al 87%, en comparación con la mamografía digital con una sensibilidad de 96,9% y una especificidad de 42,0%.
En los estudios que evaluaron la fiabilidad de la CEM en comparación con otras modalidades diagnósticas, la CEM mostró una sensibilidad del 100% y un valor predictivo negativo (VPN) del 100% en comparación con la RM (93% y 65%, p=0,04 y p <0,001, respectivamente). En comparación con la mamografía digital de campo completo (MDCC) combinada con ecografía, la CEM mostró una sensibilidad del 92,3% frente al 89,8%, p<0,05, un valor predictivo positivo (VPP) (93% frente al 88,7%, p<0,01) y fiabilidad (90,2% frente al 87%, p<0,05).
En las pacientes con contraindicaciones para la RM, tanto la clasificación por mamografía y por CEM se correlacionaron significativamente con la clasificación histopatológica. La CEM mostró una sensibilidad
Los estudios de tolerancia local tras la administración intravenosa, intraarterial e intramuscular, no mostraron reacciones adversas significativas de intolerancia.
Los estudios de genotoxicidad realizados con iopromida no mostraron potencial genotóxico.
6. DATOS FARMACÉUTICOS
6.1. Lista de excipientes
Edetato de calcio y sodio Trometamol Ácido clorhídrico (diluido al 10 %) (para ajustar el pH) Hidróxido de sodio (para ajustar el pH) Agua para preparaciones inyectables
6.2. Incompatibilidades
Este medicamento no debe mezclarse con otros, para evitar el riesgo de posibles incompatibilidades..
6.3. Periodo de validez
Antes de la apertura del envase por primera vez: 3 años. 10 horas tras la apertura del envase por primera vez.
6.4. Precauciones especiales de conservación
Antes y después de la apertura del envase por primera vez: − Conservar protegido de la luz y de los rayos X.
− No conservar a temperatura superior a 30ºC.
Para las condiciones de conservación tras la primera apertura del medicamento, ver sección 6.
6.5. Naturaleza y contenido del envase
Frasco: vidrio tipo II, incoloro. Tapón: tapón de clorobutil-caucho (tipo I, Ph. Eur.). Cápsula rebordeadora de aluminio puro con lacado interno y externo, con cubierta de cápsula coloreada de polipropileno.
Presentaciones
Envases monodosis: Frascos de 50, 75 y 100 ml. Envases multidosis: Frascos de 500 ml.
Puede que solamente estén comercializados algunos tamaños de envase.
6.6. Precauciones especiales de eliminación y otras manipulaciones
Ultravist debe ser calentado a la temperatura corporal antes de su administración.
Inspección
El medio de contraste debe inspeccionarse visualmente antes de su uso y no debe administrarse en caso de que se haya producido una alteración de su color, se evidencie la aparición de partículas en suspensión (incluyendo cristales), o en caso de que el envase esté defectuoso.
Frascos (≤ 100 ml)
La solución del medio de contraste no debe ser extraída a la jeringa, ni el frasco debe ser conectado al equipo de perfusión, hasta inmediatamente antes de la exploración.
El tapón de goma no debe ser perforado más de una vez, para evitar que grandes cantidades de micropartículas procedentes del tapón pasen a la solución. Se recomienda la utilización de cánulas de punta larga y un diámetro máximo de 18 G para perforar el tapón y extraer el medio de contraste (son particularmente apropiadas las cánulas especiales de extracción con una abertura lateral).
La solución del medio de contraste no utilizada en una exploración debe ser desechada. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
Envases de gran volumen (500 ml, únicamente para la administración por vía intraarterial o intravenosa)
La extracción múltiple del medio de contraste debe ser realizada con un equipo autorizado para la administración múltiple. Los autoinyectores/bombas no deben utilizarse en niños pequeños.
El tapón de goma del frasco no debe ser perforado más de una vez, para evitar que grandes cantidades de micropartículas procedentes del tapón pasen a la solución.
El medio de contraste debe ser administrado mediante un inyector automático, o mediante otro procedimiento aprobado que asegure la esterilidad del medio de contraste.
La conexión desde el inyector al paciente (tubo del paciente) debe ser sustituida con cada paciente, con objeto de evitar cualquier contaminación posible.
Los tubos de conexión y todas las partes desechables del sistema de inyección deben ser desechadas cuando el frasco para perfusión esté vacío o 10 horas después de la primera apertura del envase.
Es imprescindible seguir las instrucciones complementarias suministradas por los fabricantes de los respectivos materiales empleados.
El contraste que permanece en el envase de Ultravist abierto, debe ser desechado diez horas después de que se haya abierto el envase. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Hispania, S.L. Avda. Baix Llobregat, 3- 08970 Sant Joan Despí (Barcelona) España