



UNIVERSIDAD NACIONAL DE MOQUEGUA
Escuela Profesional De Ingeniería Ambiental
“CLASIFICACIÓN DE MICROORGANISMOS ”
INFORME
PRESENTADA POR:
Mireya Diana Castillo Astete
DOCENTE:
DR. HEBERT HERNAN SOTO GONZALES
Ilo – PERÚ
2026



Prepara tus exámenes y mejora tus resultados gracias a la gran cantidad de recursos disponibles en Docsity

Gana puntos ayudando a otros estudiantes o consíguelos activando un Plan Premium

Prepara tus exámenes
Prepara tus exámenes y mejora tus resultados gracias a la gran cantidad de recursos disponibles en Docsity
Prepara tus exámenes con los documentos que comparten otros estudiantes como tú en Docsity
Encuentra los documentos específicos para los exámenes de tu universidad
Estudia con lecciones y exámenes resueltos basados en los programas académicos de las mejores universidades
Responde a preguntas de exámenes reales y pon a prueba tu preparación
Consigue puntos base para descargar
Gana puntos ayudando a otros estudiantes o consíguelos activando un Plan Premium
Comunidad
Pide ayuda a la comunidad y resuelve tus dudas de estudio
Ebooks gratuitos
Descarga nuestras guías gratuitas sobre técnicas de estudio, métodos para controlar la ansiedad y consejos para la tesis preparadas por los tutores de Docsity
CODIGOS GENETICOS DE DIVERSOS ADN DE MICROORGANISMOS
Tipo: Apuntes
1 / 7

Esta página no es visible en la vista previa
¡No te pierdas las partes importantes!
El análisis molecular de secuencias nucleotídicas constituye una herramienta esencial en los estudios de caracterización e identificación microbiana, especialmente cuando se busca evaluar la diversidad genética y establecer relaciones filogenéticas entre diferentes organismos. En este contexto, el uso de software bioinformático especializado se ha consolidado como un componente fundamental para el procesamiento, edición y comparación de secuencias obtenidas por técnicas de biología molecular, tales como la amplificación y secuenciación de regiones ITS o genes ribosomales.
Entre estas herramientas, GALAXY se destaca por ser un editor de secuencias versátil y ampliamente utilizado en entornos académicos y de investigación. Su plataforma permite realizar alineamientos múltiples, corregir errores de lectura, visualizar regiones conservadas y preparar datos para análisis filogenéticos posteriores. En el estudio analizado, este software desempeñó un papel crucial en la edición y alineamiento de las secuencias parciales obtenidas, permitiendo compararlas con bases de datos internacionales y facilitando la inferencia de relaciones evolutivas mediante métodos como máxima verosimilitud.
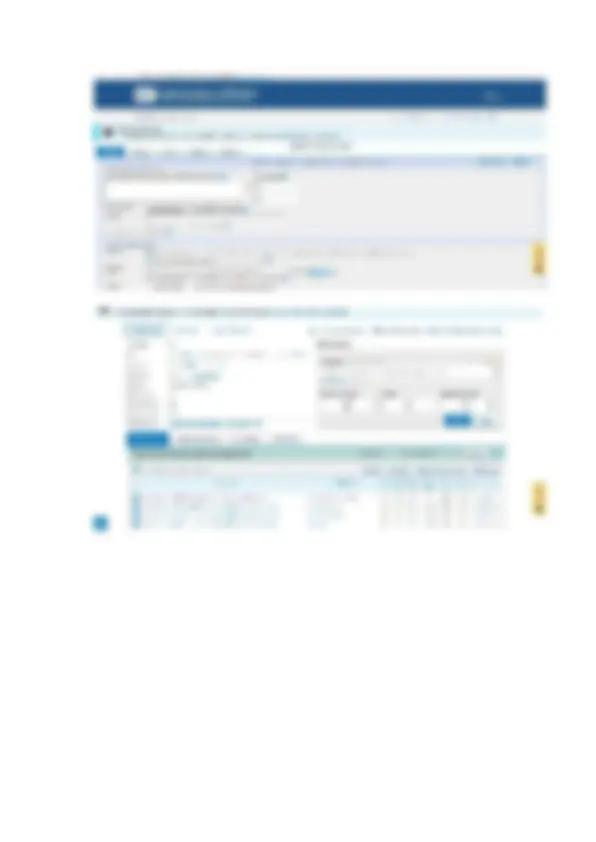
-Identificación de secuencias homólogas mediante BLAST
Se ingresó a la plataforma BLASTn del NCBI: o En el apartado “Enter Query
Sequence”, se pegó la secuencia FASTA.
Figura 3: Identificación de secuencias genética en plataforma BLASTn del NCBI.
● https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=B lastSearch&LINK_LOC=blasthome ● https://galaxy-main.usegalaxy.org/ IV. PAGINAS COMPARTIDAS ●