Baixe Material complementar - Zaha e outras Manuais, Projetos, Pesquisas em PDF para Biologia molecular, somente na Docsity!
3.1 Compactação de genomas
virais
Os genomas virais devem ser acomodados de maneira or- ganizada em um capsídeo , formando uma partícula vi- ral completa ( vírion ). O capsídeo é constituído por uma ou mais proteínas codificadas por genes virais, e o seu volume interno é apenas um pouco maior que o volume do genoma nele compactado. Os vírus são heterogêneos, quanto à natureza (de DNA ou RNA) ou à forma (circular, linear, de fitas simples ou dupla etc.) de seus genomas e os mecanismos de montagem dos capsídeos também va- riam bastante conforme o tipo de vírus.
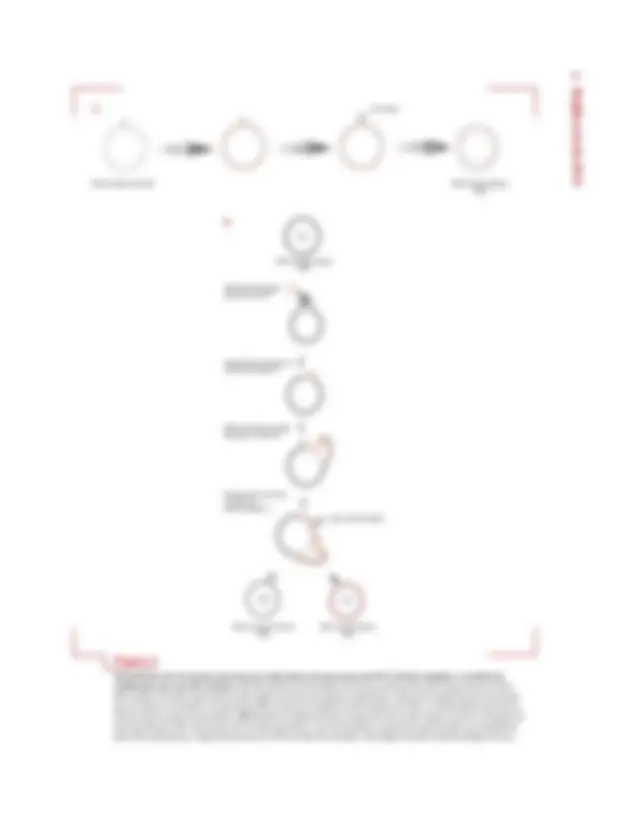
Pode-se dividir os mecanismos para a compactação de genomas virais no interior de capsídeos em dois tipos básicos. No primeiro mecanismo comum entre vírus fi- lamentosos , as proteínas do capsídeo são montadas ao redor do ácido nucleico, que é condensado à medida que o processo progride. No segundo mecanismo, comum en- tre vírus com capsídeos esféricos (com simetria ico- saédrica), o capsídeo é montado a partir dos seus compo- nentes proteicos como uma cápsula vazia, sendo o ácido nucleico posteriormente inserido e compactado no seu interior. Há, contudo, inúmeras variantes para cada um desses mecanismos, serão apresentados alguns exemplos de processos de compactação de genoma e montagem de capsídeos que ocorrem em vírus procarióticos (bacterió- fagos) e eucarióticos.
Em casos de vírus com genomas de RNA de fita sim- ples, como o vírus do mosaico do tabaco ( TMV , de tobacco mosaic virus ), a montagem do capsídeo ocorre ao redor do genoma, formando uma partícula viral fi-
lamentosa. A montagem do capsídeo de TMV inicia em uma estrutura de grampo ( hairpin ) formada pelo RNA, chamada de centro de nucleação, e prossegue, bidire- cionalmente, até o final. A unidade inicial do capsídeo é um disco de duas camadas, cada uma delas contendo 17 subunidades proteicas idênticas. O disco é convertido a uma forma helicoidal, pela interação com o RNA que, ao longo do processo de adição de novas subunidades pro- teicas, é enrolado em um arranjo também helicoidal, no interior do capsídeo.
Capsídeos esféricos de vírus de DNA, como os bac- teriófagos � e T4 são montados em um processo em que primeiro ocorre a montagem do capsídeo e, depois, a inserção do DNA. Inicialmente, é formado um pré-cap- sídeo I, contendo um núcleo proteico em seu interior. O pré-capsídeo I, depois, é convertido em um pré-capsídeo II oco, de forma icosaédrica, no qual o DNA a seguir é inserido. A inserção do DNA provoca a expansão da par- tícula, porém mantendo a mesma forma. Finalmente, o pré-capsídeo II, preenchido com o DNA, é fechado pela adição de uma cauda proteica. Tanto em � como em T4, as moléculas de DNA a serem inseridas nos capsídeos têm de estar organizadas na forma de concatâmeros, que são séries de genomas completos associados uns aos ou- tros pelas suas extremidades.
Cada um desses bacteriófagos têm, contudo, um mecanismo próprio para determinar a quantidade de DNA a ser compactada em um capsídeo. Em �, as extre- midades de cada genoma estão “marcadas” por sequên- cias específicas, chamadas de sítios cos. O DNA de um concatâmero é inserido no pré-capsídeo II, a partir do seu sítio cos esquerdo (como definido no mapa usual do genoma de �) e o processo prossegue até que seja inseri-
Material Complementar | Capítulo 3
Cromatina
Biologia Molecular Básica
do o sítio cos direito, então, ocorre a clivagem do DNA. O complexo enzimático responsável pela inserção do DNA, cujas subunidades são codificadas pelo próprio genoma do fago, é chamado de terminase. Esse complexo possui atividades de helicase (separação das fitas do DNA), de ATPase (degradação de ATP para geração de energia) e de endonuclease sítio-específica (para clivagem do DNA), que atuam coordenadamente para que um único genoma viral seja separado do concatâmero e compacta- do no capsídeo. Nesse processo, a sequência do DNA é irrelevante, ou seja, qualquer sequência de DNA, desde que flanqueada por sítios cos e com um tamanho entre 78 e 105% (de 38 a 52 kb) do genoma de �, pode ser inse- rida no capsídeo.
No caso de T4, a inserção do DNA inicia em um sítio aleatório do concatâmero e prossegue até que a quantidade de DNA equivalente a um genoma comple- to seja inserida no capsídeo. Na realidade, a quantidade de DNA inserida é um pouco maior que a de um ge- noma completo, criando, assim, uma pequena redun- dância terminal, correspondente ao DNA adicional compactado. Esse mecanismo inclui um sistema para a avaliação precisa da quantidade de DNA a ser inserida no capsídeo, dependendo tanto de sequências específi- cas como do número de cópias de sequências homólo- gas presente.
Mecanismos mais complexos de compactação de ge- nomas virais são encontrados em vírus com genomas segmentados , isto é, formados por mais de uma molé- cula de ácido nucleico, como reovírus e os vírus da gri- pe ( Influenza ). Em reovírus , um genoma constituído por 10 segmentos de RNA de fita dupla deve ser com- pactado no capsídeo. Nesse caso, sequências específicas dos segmentos virais são necessárias para garantir que uma cópia de cada um dos segmentos seja selecionada no processo de compactação, de modo a comporem um genoma completo no interior de cada capsídeo. Os ví- rus da gripe , por sua vez, possuem segmentos de RNA de fita simples, que constituem o genoma (vRNA), ini- cialmente compactado em um arranjo helicoidal com proteínas estruturais (NP e NEP) e com uma RNA- -polimerase. Esse ribonucleocapsídeo (vRNP) é, então, envolvido por uma proteína de matriz (M1) de origem viral. Por fim, é agregado um envoltório viral, contendo lipídeos do hospedeiro (na forma de uma bicamada) e glicoproteínas virais transmembranares (HA, NA e M2). O envoltório só é agregado no momento da exportação do vírion para fora da célula, de modo que partículas virais completas nunca são encontradas no interior da célula hospedeira.
Os genomas segmentados de alguns vírus tam- bém podem ser compactados em diferentes capsídeos. Um exemplo disso é o vírus do mosaico da alfafa ( AMV , de alfafa mosaic virus ), cujo genoma é cons- tituído por quatro moléculas de RNA de fita simples diferentes, cada uma compactada independentemente em uma partícula viral distinta. Assim, o sucesso da infecção com o AMV depende da presença simultânea de quatro tipos de partículas na célula hospedeira. No processo de compactação, são pré-formados quatro tipos de capsídeos que, apesar de constituídos por su- bunidades idênticas de uma mesma proteína, possuem tamanhos distintos. Cada um dos quatro tipos de mo- léculas do RNA viral é então inserido e compactado em um capsídeo com o tamanho adequado ao seu, auxilia- do pelas proteínas de montagem. Essas proteínas são codificadas pelo genoma do AMV e necessárias para a formação dos capsídeos, embora, estruturalmente, não façam parte de qualquer um deles.
Mecanismos de compactação do material genético viral, similares aos da cromatina eucariótica, são também encontrados em alguns vírus. Na morfogênese do vírus símio 40 ( SV40 , de simian virus 40 ), o DNA viral (de fita dupla circular) é primeiro compactado por histonas da célula hospedeira em uma estrutura de cromatina composta por 24 nucleossomos. Esse minicromossomo viral é, depois, incluído em um capsídeo formado pelas proteínas virais VP1, VP2 e VP3. No adenovírus , o DNA é compactado por subunidades de uma única proteína de capsídeo, sem a participação de histonas. Entretanto, quando o seu DNA passa a um estado funcional na célula, as subunidades da proteína do capsídeo são removidas e substituídas por histonas, formando nucleossomos. Ape- sar das semelhanças estruturais entre os nucleossomos de genomas virais e os nucleossomos de cromossomos das células hospedeiras, é importante salientar que há al- gumas diferenças importantes, principalmente no que diz respeito a modificações pós-traducionais nas histonas. Essas diferenças permitem que a cromatina viral tenha um comportamento funcional (de replicação e de trans- crição) independente e distinto daquele da cromatina da célula hospedeira.
3.2 Nomenclatura das
proteínas da Tabela 3.
As proteínas estão designadas de acordo com o nome dos respectivos genes (AsnC, EbpfC, Lsr2, MukB, MvaT e StpA) ou por abreviaturas. Abreviaturas:
Biologia Molecular Básica
segmentos de cromatina por uma DNase é correlaciona- do com um menor grau de compactação da estrutura e vice-versa. Regiões de menor compactação, como na fi- bra de 10 nm, estão mais acessíveis para interação com proteínas que se ligam ao DNA e, por isso, são chamadas de configuração aberta. Nos casos mais extremos de configuração aberta, regiões totalmente livres da as- sociação com histonas constituem os chamados sítios hipersensíveis, que são os primeiros a serem clivados pela DNase. Os sítios hipersensíveis são as regiões da cromatina nas quais o DNA está mais exposto e, portanto, disponível para interação com proteínas reguladoras, envolvidas em processos como os de replicação e transcrição. Por outro lado, regiões com um maior grau de compactação, por exemplo, igual ou superior ao da fibra de 30 nm, são de-
nominadas configuração fechada. As regiões de confi- guração fechada, em princípio, restringem muito o acesso de diferentes proteínas ao DNA.
3.6 Cromossomos plumosos e
cromossomos politênicos
As mesmas fibras formadas por DNA complexado com proteínas, que constituem a eucromatina, também cons- tituem a heterocromatina e os cromossomos. Por isso, pode-se dizer que a eucromatina, a heterocromatina e os cromossomos não representam elementos distintos, mas, sim, diferentes graus de condensação do mesmo material genético. Essa informação é evidenciada pelo
Digestão parcial com DNase
Digestão mais prolongada com DNase
~400 pb
Clivagem (^) Clivagem
Clivagem
~200 pb
Octâmero de histonas da partícula central do nucleossomo
DNA de ligação
Histona de ligação
Moléculas da histona de ligação liberadas
Partículas centrais liberadas (octâmeros de histonas associados a 145-147 pb de DNA)
~10 nm de diâmetro
Figura 1
Demonstração da periodicidade da or- ganização dos nucleossomos na fibra de 10 nm da cromatina. A digestão parcial da fibra de 10 nm, com uma nuclease inespecífica (DNase), gera múltiplos de um monômero cons- tituído por um nucleossomo contendo a histona de ligação (H1 ou H5) e aproximadamente 200 pb de DNA. Na separação do DNA desses multí- meros, por eletroforese em gel (ver Capítulo 16), fragmentos com tamanhos múltiplos de 200 pb são observados. Uma digestão mais prolongada com a DNase libera as histonas de ligação e gera as partículas centrais, compostas pelo octâmero de histonas associado a 145 a 147 pb de DNA.
Cromatina
fato de a eucromatina apresentar diferentes estados de condensação, tanto durante a interfase como em deter- minadas fases da divisão celular. Estados de condensação variáveis, diferentes dos observados na heterocromatina e nos cromossomos metafásicos ou mitóticos “típicos”, são também observados em cromossomos plumosos e em cromossomos politênicos.
Os cromossomos plumosos ( Figura 2 ) repre- sentam um estado reversivelmente estendido e funcional
de cromossomos de oócitos de alguns anfíbios, nos quais o processo de meiose pode durar meses.Os cromosso- mos politênicos ( Figura 3 ), por sua vez, são produtos de até nove replicações sucessivas do DNA de um par de cromossomos homólogos, nos quais as moléculas de DNA resultantes do processo (até 1.024 cópias) não se sepa- ram. Esse tipo de cromossomo, encontrado em núcleos interfásicos de alguns tecidos larvais de dípteros, possui um grau de compactação duas vezes maior do que o do cromossomo mitótico.
0,5 mm
Quiasma Eixo
Cromômero
Alça
Alça de cromatina estendida
Cromômero formado por cromatina muito condensada
Cromatina unindo cromômeros adjacentes
10 �m
A B C
Cromátides-irmãs
Figura 2
Representação esquemática da estrutura de cromossomos meióticos plumosos de oóci- tos de anfíbios. (A) Par de cromossomos plumosos homólogos pareados (unidos por quiasmas). (B) Ampliação de uma região de um dos cromossomos do par (graus maiores e menores de condensação de cromatina nas alças estão representados por linhas contínuas e tracejadas, respectivamente). (C) Ampliação de uma pequena região do cromossomo, mostrando as cromátides-irmãs.
4.1 Os três domínios da vida
Conforme uma classificação proposta por Carl Woese, em 1990, e hoje muito aceita, os seres vivos celulares podem ser divididos em três grandes clados, denominados do- mínios. Os 3 domínios propostos originalmente por Wo- ese foram chamados de Eubacteria, Archaebacteria e Eu- karya, mas, hoje, são denominados Bacteria , Archaea e Eukarya. Os domínios Bacteria e Archaea compõem o superclado Prokarya , que inclui organismos unicelula- res caracterizados pela ausência de organelas e chamados genericamente de procariotos. O domínio Eukarya, por sua vez, inclui todos os eucariotos , um grupo heterogê- neo de organismos cujas células são caracterizadas pela presença de organelas definidas, como o núcleo, que abri- ga o genoma (ver Capítulo 5). A Figura 1 mostra a deno- minada árvore da vida , que representa a classificação dos seres vivos em três domínios.
4.2 Genomas mínimos
Um aspecto interessante a ser considerado quando se trata de tamanhos de genomas procarióticos é o conceito de genoma mínimo. Este conceito teórico definiria o menor genoma capaz de abrigar o número mínimo neces- sário de genes para a sobrevivência de uma forma de vida celular. Obviamente, o genoma mínimo de organismos celulares deve pertencer a um organismo procariótico, mas a sua definição depende também de fatores como o modo de vida e as condições ambientais. Espécies de vida livre estão expostas a ambientes variáveis, e necessi- tam de um maior número de genes (ou seja, de genomas
maiores) para adaptação e sobrevivência do que espécies parasitas ou simbióticas, as quais vivem associadas a or- ganismos hospedeiros que proporcionam nichos ecológi- cos relativamente mais estáveis. Além disso, espécies he- terotróficas, que são dependentes de substratos orgânicos externos, podem ter genomas menores por dispensarem genes associados a rotas autotróficas complexas de assi- milação de compostos orgânicos e inorgânicos.
Na natureza, o genoma conhecido que mais se apro- xima do conceito de genoma mínimo, para um organis- mo de vida livre, é o da �-proteobactéria heterotrófica marinha Pelagibacter ubique SAR11, que possui 1. kb e codifica 1.354 proteínas. Dentre as espécies parasi- tas ou simbióticas, aproxima-se mais do conceito de ge- noma mínimo a bactéria endossimbionte de células de inseto Buchnera aphidicola , que tem 422 kb e codifica apenas 362 proteínas. Outras bactérias endossimbiônti- cas, como C. rudii e Sulcia muelleri têm genomas ainda menores (de 159 kb e 245 kb, respectivamente). Entre- tanto, esses genomas são desprovidos, por exemplo, de genes associados a processos como a replicação e a trans- crição, considerados essenciais para a definição de uma forma de vida celular.
A busca por genomas mínimos utiliza diversas abor- dagens experimentais complementares in silico , in vivo e in vitro. Estudos in silico permitem, a partir de análi- ses comparativas entre genomas naturais com diferentes repertórios gênicos e da predição de rotas bioquímicas, projetar qual seria o conteúdo gênico mínimo para um sistema celular autorreplicativo. Dentre os estudos in vivo importantes para a definição de um conteúdo gê- nico mínimo para organismos celulares, pode-se citar a inativação sistemática, por mutagênese sítio-dirigida
Material Complementar | Capítulo 4
Genes e Genomas
Procarióticos
Biologia Molecular Básica
em grande escala, de cada um dos genes de espécies- -modelo bacterianas, como E. coli , Bacillus subtilis ou Mycoplasma genitalium , para a identificação daqueles efetivamente essenciais. Por fim, cabem serem citadas as abordagens mais recentes de síntese in vitro de genomas mínimos, a partir da remoção de sequências de genomas pré-existentes ou a partir da completa montagem de no- vos genomas, gene a gene. Embora ambiciosas e de difícil execução do ponto de vista técnico, algumas iniciativas para a produção de genomas mínimos sintéticos já estão em andamento.
4.3 Ultraoperons em bactérias
e arqueas
Com base em dados disponibilizados por vários estudos genômicos comparativos, pode-se assumir, com elevado grau de confiança, que a organização estrutural e fun- cional em ultraoperons é prevalente tanto em bactérias
como em arqueas. No genoma de E. coli K-12, por exem- plo, foram preditos 158 ultraoperons, que, em conjunto, abrangem mais de 1.800 genes (aproximadamente 40% do repertório gênico da espécie). Cada um desses ultra- operons evidencia um conjunto de genes cuja expressão é coordenada, e cujos produtos são componentes de um mesmo processo biológico ou de um pequeno número de processos biológicos que funcionam de maneira integra- da. Muitos dos ultraoperons preditos em E. coli corres- pondem a rotas metabólicas ou outros processos bioló- gicos altamente relacionados, corroborando a vinculação funcional dos genes envolvidos. Esse é o caso, por exem- plo, do ultraoperon de proteínas ribossômicas, que inclui componentes das maquinarias de transcrição e tradução, integradas no processo de expressão gênica. Outros ca- sos, incluindo genes sem uma associação funcional mais direta à luz do conhecimento atual, são considerados como representativos de processos biológicos ainda a se- rem elucidados.
Em arqueas, o número de ultraoperons até agora descritos é menor do que o de bactérias. Por exemplo,
Proteobacteria
Actinobacteria
Planctomycetes Spirochaetes
Fibrobacteres Chlorobi Bacteroidetes
Firmicutes
Acidobacteria
Cyanobacteria Deinococcus-Thermus Chloroflexi Aquificae Thermotogae Fusobacteria Chlamydiae
Bacteria
Archaea
Eukarya
Crenarchaeota
Euryarchaeota
Nanoarchaeota
Excavata
Rhizaria
Chromalveolata
Plantae
Amoebozoa
Opisthokonta
Figura 1
Árvore filogenética mostrando a divisão dos seres vivos em três domínios (Bacteria, Archaea e Eu- karya). Os domínios procarióticos (Bacteria e Archaea) e o eucariótico (Eukarya) estão representados pelos filos e supergrupos que os compõem. Os clados estão distribuídos na árvore conforme comparações in silico , com base nas se- quências genômicas completas de espécies representativas de cada um deles. Por simplificação, as extensões dos ramos na árvore não correspondem às distâncias evolutivas exatas entre filos, supergrupos e domínios. Árvore estruturada conforme dados filogenômicos de Ciccarelli et al. (2006).
Biologia Molecular Básica
a uma frequência elevada de rearranjos cromossômicos associados, mas os mecanismos moleculares associados a esses determinantes de instabilidade ainda não foram elucidados.
Contração de genomas
Em procariotos, a contração genômica geralmente ocorre como uma resposta adaptativa a um ambiente mais sim- ples e/ou estável que o ocupado anteriormente por uma espécie (ou por uma população isolada de indivíduos de uma espécie). Esse foi o caso de B. aphidicola , discutido acima, que teve seu genoma reduzido ao evoluir de uma condição ancestral de vida livre para uma condição de endossimbionte. Em processos de contração genômica como esse, uma redução de tamanho populacional asso- ciada a uma mudança de hábitat e/ou de nicho ecológico leva à fixação de mutações e à expansão do número de cópias de elementos transponíveis, pois torna a seleção purificadora ineficiente e favorece a deriva genética. Com isso, muitos genes são inativados por mutações e/ou por inserção de elementos transponíveis. Além disso, os ele- mentos transponíveis, por estarem repetidos no genoma, também aumentam a probabilidade de ocorrência de re- arranjos cromossômicos. São esses rearranjos cromossô- micos que, em última análise, acabam por efetivar a con- tração genômica, levando à deleção de algumas regiões do genoma e à perda de genes.
O processo de perda de genes na contração genômica é geralmente irreversível. As mutações acumuladas não podem ser corrigidas e o pequeno tamanho populacio- nal torna improvável a recuperação de genes através de eventos de transferência gênica horizontal. A contração genômica é adaptativa porque a maioria dos genes perdi- dos (inicialmente por inativação funcional e, depois, por deleção) corresponde a funções necessárias apenas em hábitats ou modos de vida prévios da espécie, ao passo que os genes que são preservados são os que viabilizam a sua sobrevivência e multiplicação nas condições ambien- tais vigentes.
A ocorrência de processos de contração genômica em andamento pode ser evidenciada com base em “pis- tas” presentes nos genomas envolvidos, como a presen- ça de pseudogenes (indicativa de inativação gênica), a ausência de ortólogos de genes presentes em espécies aparentadas (indicativa de perda de genes ) e/ou o ele- vado número de cópias de elementos transponíveis (in- dicativa de falta de seleção contrária à expansão desses elementos). Essas linhas de evidência são encontradas, por exemplo, no genoma de Bordetella pertussis , um patógeno pulmonar – hoje restrito a seres humanos – que evoluiu de uma espécie ancestral (similar a Borde- tella bronchiseptica ) capaz de infectar também outras espécies de mamíferos. Em comparação com o genoma de B. bronchiseptica (5,7 Mb), o genoma de B. pertussis (4 Mb) perdeu mais de 20% de seu DNA (por deleções) e quase 10% dos genes remanescentes foram inativa- dos, principalmente devido a inserções de um elemento transponível, que está presente em mais de 240 cópias.
Como não há evidência de qualquer aquisição de genes compensatória por B. pertussis , esse é considerado um caso típico de contração genômica em andamento.
Expansão de genomas
Certas combinações de fatores genéticos e ecológicos também podem resultar em situações nas quais o genoma de uma espécie procariótica tende a expandir, com a in- corporação estável de novos genes. A expansão genômica depende de sucessivos eventos de duplicação gênica e/ou de transferência gênica horizontal, mas as novas sequên- cias assim adquiridas só podem ser fixadas em condições nas quais há forte pressão seletiva favorável à fixação e/ ou a pressão seletiva contrária a ela não é efetiva. Como são raros os casos nos quais genes recém-duplicados ou recém-adquiridos por transferência gênica horizontal conferem vantagens adaptativas pronunciadas (passíveis de forte seleção positiva), é mais comum que a expansão genômica esteja associada a condições ambientais e de dinâmica populacional nas quais as pressões para eli- minação das novas sequências estão enfraquecidas. As- sim, processos de expansão genômica mais significativos geralmente ocorrem em espécies que ocupam hábitats complexos e variáveis, e que vivem em pequenas popu- lações ou passam, às vezes, por situações de redução do tamanho populacional (chamadas de “gargalos popula- cionais”). Isso acontece porque, em ambientes complexos e variáveis, é maior a probabilidade de genes recém-ad- quiridos virem a ter algum valor adaptativo, e, em peque- nas populações, o relaxamento da seleção purificadora e a ação da deriva genética permitem a fixação de novas sequências adquiridas, independentemente da adaptabi- lidade conferida por elas.
Genomas expandidos são comuns entre espécies pro- carióticas de diferentes grupos taxonômicos que vivem no solo, o que denota a importância do ambiente (com- plexo e variável) como um dos determinantes de proces- sos de expansão genômica. Um exemplo típico é o de S. coelicolor , uma actinobactéria de solo que forma colônias multicelulares diferenciadas, inclusive com a formação de um micélio aéreo e esporos. No genoma de S. coelico- lor A3(2), de 8,7 Mb, são encontrados 7.865 genes , quase o dobro do número encontrado em M. tuberculosis , que pertence à mesma ordem (Actinomycetales) de actino- bactérias. Esse repertório gênico, enriquecido durante o processo de expansão genômica, inclui genes envolvi- dos no transporte e degradação de nutrientes extracelu- lares, o que permite a S. coelicolor aproveitar a matéria orgânica em decomposição proveniente de praticamen- te qualquer tipo de organismo, como genes reguladores, necessários à resposta a diferentes tipos de estresse e a estímulos ambientais, e genes codificadores de enzimas envolvidas na produção de vários metabólitos secundá- rios, inclusive antibióticos e pigmentos. Além disso, há muitos genes associados ao desenvolvimento miceliano e à esporulação.
Muitos pares de genes duplicados (parálogos) podem ser identificados, demonstrando a relevância de eventos
Genes e Genomas Procarióticos
de duplicação gênica no processo de expansão genômica S. coelicolor. Dentre os pares de parálogos identificáveis, vários codificam formas alternativas (isoformas) de pro- teínas expressas diferencialmente em células vegetativas, em células micelianas ou em esporos, demonstrando que, paralelamente à diversificação do repertório proteico pro- porcionada pela duplicação, evoluíram mecanismos de regulação da expressão gênica associados à diferenciação
celular, raros em procariotos. Além disso, pelo menos 20 segmentos do genoma de S. coelicolor foram provavel- mente adquiridos por transferência gênica horizontal, ten- do o maior deles 153 kb e 148 genes. Dentre os genes assim adquiridos há vários de origem plasmidial, sugerindo que pelo menos alguns dos processos de transferência horizon- tal ocorridos na evolução de S. coelicolor foram mediados por elementos (plasmídeos) conjugativos e integrativos.
Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B, Bork P. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311(5765):1283-7. Keeling PJ. Genomics. Deep questions in the tree of life. Science. 2007;317(5846):1875-6.
Morell V. Life’s last domain. Science. 1996;273(5278):1043-5. Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87(12):4576-9.
Leituras recomendadas
Biologia Molecular Básica
sar desses exemplos extremos, há uma frequência muito maior de íntrons com tamanhos entre 50 pb e 2 kb para a maioria das espécies eucarióticas já estudadas quanto a esse aspecto. Parece haver uma tendência de aumento da frequência de íntrons com maior tamanho à medida que se avança na escala evolutiva. Essa questão pode ser demonstrada pela comparação de medianas, pois, como medidas de tendência central, elas evitam os desvios às vezes significativos que ocorrem nas médias de algumas espécies pela ocorrência de alguns poucos íntrons de ta- manho muito grande. Assim, considerando-se as media- nas dos tamanhos dos íntrons em cada genoma, valores crescentes são observados quando comparamos, por exemplo, Schizosaccharomyces pombe (52 pb), C. ele- gans (65 pb), D. melanogaster (78 pb), A. thaliana ( pb), arroz (145 pb), Ciona intestinalis (um cordado inver- tebrado, 318 pb), galinha (846 pb) e o homem (1,4 kb).
Ao contrário do que ocorre com os íntrons (ampla variação de tamanho), o tamanho dos éxons dos ge- nes eucarióticos interrompidos varia, via de regra, den- tro de limites restritos. O tamanho dos éxons é, em geral, pequeno, com a grande maioria variando entre 50 e 500 pb. Para o tamanho de éxons, as médias são também des- viadas pela ocorrência de alguns poucos éxons maiores em algumas espécies, sendo então as medianas, como no caso dos íntrons, mais informativas para efeitos com- parativos. Assim, considerando-se as medianas, valores muito próximos são observados para espécies eucarióti- cas de diferentes supergrupos e filos, como S. pombe ( pb), Plasmodium falciparum (109 pb), C. elegans ( pb), D. melanogaster (204 pb), A. thaliana (153 pb), C. intestinalis (129 pb) e o homem (124 pb).
Os éxons, que incluem as sequências codificadoras dos produtos gênicos de eucariotos, não são os principais
Apicomplexa Ciliophora Cryptophyta Dinoflagellata Haptophyceae Stramenopiles
Diplomonadida Euglenozoa Heterolobosea Hypermastigia Jakobidae Kinetoplastida Malawimonadidae Oxymonadida Retortamonadidae Trichomonadida
Cercozoa Foraminifera Radiolaria
Archamoebae Euamoebida Lobosea Mycetozoa
Metazoa Choanoflagellida Chytridiomycota Dikarya Ichthyosporea Microsporidia Nucleariidae Eukarya
Bacteria Archaea
Cholorophyta Embryophyta Glaucophyta Rodophyta
Excavata
Rhizaria
Chromalveolata
Plantae
Amoebozoa
Opisthokonta
Figura 1
A árvore filogenética eucariótica. Nesta representação esquemática, o comprimento dos ramos da árvore é arbitrário, não tendo qualquer correlação com distâncias evolutivas. Dentro de cada supergrupo (balões em cor de rosa), são listados grupos taxonômicos de espécies representativas. Os grupos taxonômicos são de diferentes hierarquias, podendo variar de famílias a filos ou outros táxons de ordem superior, e, em cada supergrupo, estão ordenados alfabeticamente. Os grupos ta- xonômicos que incluem os eucariotos mais conhecidos estão indicados por retângulos em magenta: Metazoa corresponde aos animais; Dikarya corresponde aos fungos típicos (ascomicetos e basidio- micetos); e Embryophyta corresponde aos vegetais terrestres (plantas vasculares e briófitas).
Genes e Genomas Ecarióticos
determinantes do tamanho dos genes desses organismos. Em relação ao tamanho, o número e o tamanho de íntrons são os determinantes mais importantes. Assim, como há tendências de aumento, tanto do número de íntrons por gene como do tamanho médio dos íntrons, à medida que se avança na escala evolutiva, os genes de eucariotos mais complexos tendem a ser maiores, que os genes de euca- riotos mais simples.
5.3 Famílias multigênicas da
actina e da globina
A família de genes de actina é um bom exemplo de família multigênica ubíqua em eucariotos. As actinas são proteínas bastante conservadas (as sequências de aminoá- cidos de actinas de animais e plantas diferem em apenas 25 a 30%) e o número de genes que compõe a família que as codifica é variável em diferentes espécies. Em levedu- ras ( S. cerevisiae e S. pombe ) e no ciliado Tetrahymena thermophila (Alveolata), há apenas um gene codificando actina, mas mesmo outras espécies eucarióticas também simples, como Oxytricha fallax e Paramecium tetraure- lia (Alveolata) e Entamoeba hystolitica e Dictyostelium discoideum (Amoebozoa), já apresentam de 3 a mais de 10 genes na família. Entre metazoários, o número de ge- nes da família é relativamente pequeno em C. elegans ( genes ) em D. melanogaster (6 genes ) e no ouriço-do-mar Strongylocentrotus purpuratus (5 genes ), mas tende a aumentar em vertebrados, com aves e anfíbios tendo em torno de 10 genes e mamíferos chegando a ter algumas poucas dezenas (30 no homem). Em vegetais superiores, como soja, milho e arroz, há pelo menos de 6 a 10 genes de actina. Quanto à localização, a maioria das espécies estudadas possuem seus genes de actina dispersos pelo genoma, mas algumas espécies, como C. elegans e S. pur- puratus , têm pelo menos alguns dos seus genes de actina agrupados em uma mesma região cromossômica.
Um aspecto interessante da família multigênica da actina é a sua diversificação funcional em eucariotos mais complexos. Em insetos, como moscas do gênero Droso- phila , são observados alguns genes codificando isoformas de actina musculares especializadas (larvais ou adultas), e outros codificando actinas não musculares, generica- mente chamadas de citoplasmáticas. A diversificação funcional é ainda mais exacerbada em vertebrados, onde são identificadas pelo menos 6 isoformas diferentes, duas citoplasmáticas (actinas � e �), duas de músculos estriados (actinas �-esquelética e �-cardíaca) e duas de músculos lisos (actinas �-vascular e �-entérica). Os genes das isoformas citoplasmáticas são expressos em todos os tipos celulares, e os das isoformas musculares são expres- sos em órgãos e tipos de músculo específicos.
A família de genes de globina de vertebrados, por sua vez, é um bom exemplo de família multigênica que se expandiu e diversificou ao longo de uma linhagem eucari- ótica específica. Em vertebrados não mandibulados (como
as lampreias), a hemoglobina é monomérica e codificada por um único gene de (hemo)globina. Entretanto, graças a uma duplicação gênica que teria ocorrido entre 500 e 570 milhões de anos e deu origem aos genes de globina � e �, todos os vertebrados mandibulados (superclasse Gnathostomata) têm hoje hemoglobinas tetraméricas, formadas por duas globinas de tipo � e duas de tipo �. Inicialmente, os genes ancestrais de globina � e � dupli- cados mantiveram-se próximos no genoma, em um tipo de organização que foi preservada em peixes teleósteos e anfíbios. Na linhagem evolutiva que deu origem a aves e mamíferos, ocorreu uma separação dos genes de globina � e � em cromossomos diferentes, situação preservada nas espécies modernas dessas duas classes de vertebrados.
Ao longo da evolução dos vertebrados, eventos adi- cionais de duplicação foram acontecendo em linhagens específicas, dando origem a novos genes de globina � e � funcionais e também a alguns pseudogenes. O número e a ordem dos genes em cada subfamília (� ou �) podem va- riar de espécie para espécie, sendo que, em alguns casos, um lócus ocupado por um pseudogene em uma espécie pode corresponder a um gene funcional em outra. É in- teressante notar que exigências associadas a mecanismos específicos de expressão levaram à manutenção dos pará- logos gerados em cada subfamília de uma mesma região cromossômica, determinando a formação de agrupa- mentos (do inglês, clusters ) de genes com padrões defi- nidos de expressão espacial e temporal. Assim, distintas isoformas de hemoglobina são expressas na linhagem celular eritrocitária em diferentes estágios do desenvolvi- mento de vertebrados superiores.
No homem, o agrupamento de genes de globina � está posicionado no cromossomo 16, e o agrupamento de genes de globina � está no cromossomo 11. A organização de cada um desses agrupamentos no genoma humano e de outros primatas superiores está representada na Figura 2. O agrupamento � ocupa aproximadamente 50 kb e inclui cinco genes funcionais – �, dois genes �, � e � – e um pseu- dogene ( �1). Os dois genes � diferem em apenas um nu- cleotídeo, o que determina a alteração de um único ami- noácido nas proteínas correspondentes. O agrupamento � é menor, estendendo-se por mais ou menos 28 kb. Esse agrupamento inclui um gene funcional, um pseudogene , dois genes �, dois pseudogenes � e um gene. Os genes � são cópias não alélicas , isto é, cópias idênticas entre si, presentes em um mesmo cromossomo. A partir da ex- pressão diferencial dos genes desses dois agrupamentos, diferentes isoformas (tetrâmeros) de hemoglobina são ge- radas nos estágios de desenvolvimento embrionário ( 2 � 2 , � 2 � 2 e 2 � 2 ), fetal (� 2 � 2 ) e pós-natal (� 2 � 2 e � 2 � 2 ).
5.4 Estimativas de tamanho de
genomas eucarióticos
A maneira mais precisa para determinar o tamanho do genoma de uma espécie é o seu sequencimento com-
Genes e Genomas Ecarióticos
Onde: x é o tamanho do genoma desconhecido e 4,6 x 10 6 é o tamanho do genoma de E. coli (em pares de bases).
Em cinéticas de renaturação de genomas eucarióti- cos mais complexos, componentes reassociativos distin- tos podem ser identificados, correspondendo a cada um deles um valor de C 0 t (^) ½ distinto ( Figura 3 ). Valores de C 0 t (^) ½ menores correspondem a frações do genoma mais ricas em sequências repetidas, que se reassociam mais rápido. Isso é uma consequência lógica, partindo do
fato de que a presença de um número maior de cópias de uma determinada sequência aumenta a probabilida- de de encontro e pareamento das fitas complementares correspondentes e, em consequência, resulta no aumen- to da velocidade (e redução do tempo) do processo de renaturação de uma fração do genoma contendo DNA repetitivo. Assim, a partir de experimentos comparativos de cinética de renaturação, pode não só ser estimado o tamanho total de um genoma eucariótico, como também os tamanhos de seus componentes de sequência com di- ferentes graus de reiteração.
Besansky NJ, Powell JR. Reassociation kinetics of Anopheles gambiae (Diptera: Culicidae) DNA. J Med Entomol. 1992;29(1):125-8. Bolstad AI. Sizing the Fusobacterium nucleatum genome by pulsed-field gel electrophoresis. FEMS Microbiol Lett. 1994;123(1-2):145-51. Dolezel J, Bartos J. Plant DNA flow cytometry and estimation of nuclear genome size. Ann Bot. 2005;95(1):99-110. Greilhuber J. Cytochemistry and C-values: the less- well-known world of nuclear DNA amounts. Ann Bot. 2008;101(6):791-804. Hackett JD, Yoon HS, Li S, Reyes-Prieto A, Rümmele SE, Bhattacharya D. Phylogenomic analysis supports the monophyly of cryptophytes and haptophytes and the Association of Rhizaria with Chromalveolates. Mol Biol Evol. 2007;24(8):1702-13.
Hampl V, Hug L, Leigh JW, Dacks JB, Lang BF, Simpson AG, et al. Phylogenomic analyses support the monophyly of Excavata and resolve relationships among eukaryotic "supergroups". Proc Natl Acad Sci U S A. 2009;106(10):3859-64. Keeling PJ, Burger G, Durnford DG, Lang BF, Lee RW, Pearlman RE, et al. The tree of eukaryotes. Trends Ecol Evol. 2005;20(12):670-6. Neafsey DE, Palumbi SR. Genome size evolution in pufferfish: a comparative analysis of diodontid and tetraodontid pufferfish genomes. Genome Res. 2003;13(5):821-30. Wilhelm J, Pingoud A, Hahn M. Real-time PCR-based method for the estimation of genome sizes. Nucleic Acids Res. 2003;31(10):e56. Yoon HS, Grant J, Tekle YI, Wu M, Chaon BC, Cole JC, et al. Broadly sampled multigene trees of eukaryotes. BMC Evol Biol. 2008;8:14.
Leituras recomendadas
10 -4 10 -3 10 -2 10 -1 1 10 102 103 104
75
50
25
100
C 0 t
Fração reassociada (%) A
C
B
Figura 3
Representação gráfica da cinética de renaturação de um ge- noma eucariótico com três componentes reassociativos dis- tintos. No gráfico, o eixo das abcissas representa os valores de C 0 t (^) ½, e o eixo das ordenadas representa a fração renaturada da amostra de DNA genômico. A cinética de renaturação gera uma curva complexa, poden- do ser separada em três segmentos sigmoidais (A, B e C), sombreados em cinza. O componente A corresponde a sequências altamente repe- titivas, que se reassociam com rapidez; o componente C corresponde a sequências únicas (não repetidas), que se reassociam mais lentamente; e o componente B corresponde a sequências repetitivas moderadas, que se reassociam com uma velocidade intermediária a dos componentes A e C. As interseções das linhas pontilhadas definem os pontos de inflexão das curvas sigmoides e, consequentemente, os valores de C 0 t (^) ½, de cada um dos três componentes (indicados por setas)
Replicação do DNA
Material Complementar | Capítulo 6
6.1 Tipos de DNA-polimerases
Existem, até o momento, seis famílias de DNA-polimera- ses separadas conforme a similaridade de sequências. As DNA-polimerases envolvidas no processo de replicação, tanto de organismos procarióticos como eucarióticos, são classificadas nas famílias A, B e C. A DNA-polimerase III de E. coli e a DNA-polimerase � de eucariotos pertencem à família C e as DNA-polimerases � e � de eucariotos per- tencem à família B. A DNA-polimerase I de E. coli , neces- sária também para a replicação, é um representante da família A.
As outras DNA-polimerases de E. coli são classifi- cadas nas famílias X e Y. DNA-polimerases pertencen- tes à família X e Y participam dos eventos de reparação, principalmente nos processos em que ocorrem lesões no DNA, sendo responsáveis pela síntese da região conten- do o dano. No entanto, DNA-polimerases pertencentes à família B também podem estar envolvidas com estes mecanismos. A principal diferença funcional entre as DNA-polimerases das famílias X e Y está relacionada à fidelidade. DNA-polimerases da família X, como a DNA- -polimerase II de E. coli , apresentam uma fidelidade moderada durante a síntese do DNA, sendo inferior à fidelidade das replicases. No entanto, a síntese de DNA realizada pelas DNA-polimerases da família Y ocorre com número considerável de mutações, pois essas enzi- mas não contêm a atividade de exonuclease 3' → 5' para correção de erro.
DNA-polimerases da família Y são encontradas em bactérias, arqueas e eucariotos, sendo sempre relacio- nadas à síntese para o reparo de DNA contendo dano.
Neste grupo, encontram-se as DNA-polimerases IV e V de E. coli e também, as DNA-polimerases , , � e Rev de organismos eucarióticos. Apesar de não existir simi- laridade de sequência entre as DNA-polimerases dessa família com as DNA-polimerases replicativas, as DNA- -polimerases da família Y apresentam uma estrutura muito similar à das DNA-polimerases associadas à repli- cação. Estudos cristalográficos das DNA-polimerases da família Y demonstraram que elas apresentam uma con- formação em forma de mão humana, similar à descrita para a DNA-polimerase I de E. coli. Entretanto, além da estrutura de “palma da mão, dedos e polegar”, DNA-po- limerases da família Y contêm um dedo adicional, que parece estar relacionado com a especificidade de subs- trato e à processividade. Esta estrutura adicional, deno- minada “dedo pequeno”, interage com a subunidade � da DNA-polimerase III, permitindo, dessa forma, que essas polimerase tenham acesso ao DNA durante even- tos de reparo posteriores à replicação. Um aspecto rele- vante é o de que a processividade das DNA-polimerases da família Y aumenta com a ligação à subunidade � da DNA-polimerase III, contribuindo para modular sua ati- vidade.
6.2 Mecanismos de círculo
rolante para replicação de
plasmídeos
Diferentes plasmídeos utilizam mecanismos de círculo rolante para sua replicação. Plasmídeos pequenos, mul- ticópia (com número de cópias por célula variado), tan-
Replicação do DNA
A
B
DNA de fita simples DNA de fita dupla (RF)
Primase Polimerase
Iniciador
SSBSSB
DNA de fita dupla (RF)
DNA de fita simples (SS)
RF
SS RF
DNA de fita dupla (RF)
5'
Quebra da ligação pela proteína A
Ligação da proteína A na extremidade 5'
Nova fita de DNA
Deslocamento da fita ligada à proteína A
Síntese da nova fita a partir da extremidade 3'
3'
3'
Figura 2
Mecanismo de formação das formas replicativas de genomas de DNA de fita simples e modelo de replicação por círculo rolante. (A) Mecanismo da formação das formas replicativas (RF) de genomas de DNA fita simples. Os DNAs apresentam uma região em forma de grampo originando o sítio de reconhecimento da primase para a síntese do iniciador. As proteínas SSB recobrem as regiões de fita simples do DNA e a DNA-polimerase inicia a síntese da fita a partir do iniciador. (B) Modelo de replicação pelo mecanismo de círculo rolante. Ocorre a clivagem de uma das fitas do DNA das formas RF em sítio específico, e na extremidade 3' gerada são adicionados os nucleotídeos pela DNA-polimerase, originando formas de DNA circular fita simples e fita dupla (modelo do bacteriófago �X174).
Biologia Molecular Básica
região do plasmídeo F com aproximadamente 33 kb, denominada “região de transferência”, contém os ge- nes tra , sendo a maioria destes genes organizada em um único óperon transcrito como um mRNA de 32kb. Plasmídeos conjugativos apresentam sempre duas ori- gens da replicação, sendo uma delas (oriT) relacionada especificamente ao processo de conjugação. Durante a conjugação ocorre a ativação da oriT, iniciando a repli- cação por círculo rolante e transferindo para a célula receptora uma das fitas de DNA. O processo de repli- cação é similar ao descrito para os bacteriófagos ocor- rendo a transferência de DNA da fita simples. Após a passagem do DNA para a célula receptora ele então é convertido em fita dupla.
Célula hospedeira DNA de � circular
Partícula viral DNA de � linear
Circularização do DNA do bacteriófago �
Replicação por círculo rolante
A
B
3'
5'
5'
Quebra da ligação
Deslocamento da fita com extremidade 5' livre
Síntese das novas fitas de DNA
Genoma completo
5'
3'
5'
3'
cos cos
Figura 3
Modelo de replicação do bacteriófago �. O DNA de fita dupla linear da partícula viral circulariza dentro da célula hospedeira, pelos sítios cos (A). Durante a replicação, a formação de DNA linear de fita dupla ocorre por meio do mecanismo de círculo rolante gerando os concatâmeros (B).