Scarica sbobbinature di bioinformatica e più Dispense in PDF di Bioinformatica solo su Docsity!
5°lezione di Bioinformatica 28 ottobre 2014.
Genome Browser: Regolazione.
[N.B. : le porzioni di testo in corsivo sono aggiunte da me al fine di facilitare la fluidità del discorso o di aggiungere note che possono semplificare la ricerca su genome browser in punti in cui la prof ha sorvolato o è stata poco chiara o per spiegare differenze nei punti in cui slides caricate online e proiezione dei comandi live non corrispondono. Ho agigunto anche gli screenshot dei passaggi rifatti da me.] Nella lezione di oggi, diversa dalla precedente, in cui abbiamo trattato l’espressione genica, tratteremo la regolazione, di cui abbiamo parlato abbondantemente a lezione. Questa lezione servirà anche per migliorare la pratica su Genome Browser quindi non vi guiderò nel dettaglio in alcuni comandi di funzioni già analizzate la volta scorsa.
Andate su Genome Browser e cercate il gene P63 umano nella versione (“assembly”)2009:
[ google-> Genome browser-> “UCSC Genome Browser Home”-> “genome browser”(prima voce in alto a sx)-> “click here to reset”-> in “assembly” selezionare dal menu a tendina “Feb.2009(GRCh37/hg19)”-> in “serch term” digitare TP63*-> “submit”]
- N.B: Spesso uno stesso gene è stato chiamato nel corso della storia con nomi diversi oppure ad uno stesso nome possono corrispondere diversi geni, fattori ecc. Quindi per essere sicuri di andare a studiare il gene di interesse in questo caso vado in NCBI nel menu a tendina in alto a sx seleziono “gene” e nella barra a fianco scrivo “human P63”. La prima voce dei risultati della ricerca è TP “tumor protein p63” , controllo poi alla voce “aliases”(=pseudonimi) e ritrovo tra i vari pseudonimi p63 quindi so che il nome “ufficiale”del mio gene umano p63 è TP63.
In questa diapositiva è riportato lo scopo, ovvero l’uso di genome browser per identificare la regolazione trascrizionale di un gene, promotori prossimali o intronici mediante sia il codice istonico che tramite identificazione di DHS (DNAse Hypersensitive sites). Ricordate che io vi ho detto P63 ma voi dovete controllare quale è il nome reale del gene (*)! Perché se nella barra di ricerca si digita semplicemente P63 e si clicca il primo risultato potrebbe uscire una qualsiasi cosa, il gene di interesse o altro. Ricordate inoltre prima di proseguire a qualsiasi ricerca di effetturare un “reset” ( basta andare su “click here to reset” prima di inserire i parametri della ricerca ) perché potrebbero apparire cose diverse da quelle che ci servono e vorremmo. Fate uno zoom out di 1.5 volte ( in alto a dx sotto il titolo in grassetto cliccare su zoom out “1.5X” ) o fate comunque in modo di vedere il gene per esteso, sia l’inizio che una parte del promotore che una parte del 3’UTR. D: come individuo il promotore? Mi è stato chiesto se il promotore fosse indicato in qualche modo o come sia individuabile in Genome Browser, questo non è altro che la regione a monte dell’inizio della trascrizione ovvero del TSS (Transcriptional Start Site), tenendo sempre in conto che la direzione del gene è indicata con le freccette. Il promotore sarà sicuramente nelle prime 200-300 bp a monte del TSS ma non c’è una grandezza specificata, può essere più o meno ampio e sta a me decidere i parametri nel caso in cui voglia scaricarmi la sequenza del promotore. Comunque potrei trovare sequenze regolatorie anche a 5mila bp per esempio, e a quel punto la distinzione tra promotore ed enhancer è un po’ più difficile da fare. Dovreste vedere una cosa del genere: Si nota che ci sono due trascritti di lunghezza diversa, ci sono quindi due siti di inizio della trascrizione, il trascritto parte con l’esone 1. Le lunghezze diverse dei trascritti in questo caso
Le nove linee cellulari sono indicate più sotto, ogni quadratino colorato corrisponde ad una linea cellulare indicata. Ci sono linee sia endodermiche che ectodermiche che mesenchimali, ci sono le cellule staminali H1HESC che sta per Epidermal Stem Cells e anche i cheratinociti e i fibroblasti. Questa struttura, simile, l’abbiamo vista per i dati dell’espressione la scorsa volta e ci aspettiamo che i dati non siano molto dissimili, dato che trascrizione ed espressione dovrebbero andare di pari passo.
Per ogni schermata ho sempre una serie di informazioni , di comandi, che mi permettono di visualizzare i diversi dati in maniera diversa ( in riferimento ai comandi “full”=visualizza tutto “hide”=nascondi completamente, “pack”=visione compattata ecc. In linea di massima quando delle informazioni non servono, al fine di non confondersi, basta impostare “hide”, evidenziando in tal modo solo le cose che si interessa studiare. Dopo aver spuntato per ogni voce l’opzione Hide/pack/ full in fondo alla pagina cliccare “refresh all” per aggiornare.) Pur essendo solo comandi che riguardano la grafica sono importanti perché altrimenti lasciando tutto in visualizzazione completa non si riesce a focalizzarsi sulle informazioni di interesse.
( domande/risposta tra prof e studente , cfr. slide pagina precedente) D: Cosa indicano queste linee colorate? In particolare questa prima linea rossa? R: i livelli di trascrizione di linee cellulari diverse, la prima linea rossa è relativa all’espressione della prima linea cellulare. D: che trascritto si fa in questa prima linea cellulare? R: Il trascritto di P63 più lungo, quello in alto, in pratica viene trascritto tutto il gene lungo. Mentre nell’ultima linea, quella lilla ci sono picchi in corrispondenza del trascritto corto. Quindi le informazioni che otteniamo da questi dati sono: nella maggior parte delle linee cellulari si vede un bel vuoto, quindi non c’è trascrizione, nella prima linea cellulare, rossa, c’è trascrizione di tutto il trascritto lungo e non solo di quello corto, mentre in cheratinociti (lilla) il trascritto che si fa è solo quello corto e non quello lungo. -Ma a cosa serve questa cosa? Posto che questo è un gene che conosciamo benissimo, supponiamo di avere un gene che non conosciamo e vogliamo sapere in quali linee cellulari possiamo andare a studiare questo gene, magari vogliamo usare linee cellulari che esprimono il gene, oppure overesprimere il gene in linee che normalmente non lo esprimono. Quindi utilizzo queste informazioni per decidere quali linee cellulari possono essere utili per studiare questo determinato gene. Ovviamente, in questo caso, se mi interessa il trascritto corto di P63, andrò a silenziare in queste cellule ( quelle indicate dalla linea lilla: NKEH, cheratinociti) non lo andrò a silenziare ovviamente nelle altre cellule in cui il trascritto corto non c’è.
Torniamo su “ENCODE Regulation” e vediamo cos’altro possiamo fare; adesso nasconderò il trascritto che non ci interessa più perché l’abbiamo già visto, e andiamo a vedere H3K4me monometilato e H3K4me3 trimetilato e H3K27ac acetilato. ( In questo caso i dati riportati riguardano 7 linee cellulari ). Sappiamo che H3K4me3 è un segnale di inizio della trascrizione invece H3K4me1 può marcare sia enhancer attivi che inattivi. Vi ricordate i poised enhancer nelle cellule staminali?( sono enhancer inattivi ma che possono attivarsi durante il differenziamento in risposta a stimoli esterni , sono marcati da H3K27me3 e H3K9me3 ). H3K27ac lega gli enhancer effettivamente attivi. (slide non mostrata a lezione)
cheratinociti che sono le NHEK. Ricordandovi di impostare per tutti e tre come “overlay method” “none” e come “display method” “full”. _1) H3K4me1: “Encode Regulation”-> “layered H3K4me1”-> “display mode” imposto “full”-
“overlay method” imposto “none”-> dalla lista di linee cellulari spunto a mano ad una ad una le singole linee ad eccezione di NHEK che lascio selezionata -> “submit”.
- H3K4me3: “Encode Regulation”-> “layered H3K4me3”-> ..ecc. come sopra.
- H3K27ac: “Encode Regulation”-> “layered H3K27ac”-> ..ecc. come sopra. Seguendo I comandi Ottengo questi profili:_
Cosa vedo? La 2° linea lilla è il H3K4me3 che marca il sito di inizio della trascrizione all’inizio del trascritto più piccolo (infatti ho un solo picco in corrispondenza dell’inizio del trascritto piccolo che abbiamo detto essere quello tessuto specifico per cheratinociti). La 1°linea lilla riguarda il H3K4me1 in cui trovo diversi picchi che marcano sia i promotori che gli enhancer attivi e inattivi. La 3° linea lilla indica H3K27Ac in cui trovo diversi picchi corrispondenti a diversi enhancer attivi. Questa sotto è invece la slide della prof.(anche in origine le scritte sono illeggibili) in cui però non sono stati effettuati gli stessi comandi: le prime sette linee colorate riguardano H3K4me1 nelle diverse linee cellulari, dall’ottava alla quattordicesima invece riguardano H3K27Ac sempre in tutte le diverse 7 linee cellulari( quindi al contrario di come chiesto a lezione qui sono mostrate tutte le linee cellulari e non solo i cheratinociti!!! In più non è mostrato H3K4me3.). Come vedete quindi il monometilato non è tanto utile, però il H3K27ac non solo mi fa vedere il promotore ma mi fa vedere dei picchi molto interessanti all’interno. In una delle prime lezioni vi ho raccontato il lavoro che abbiamo fatto in laboratorio per identificare gli enhancer di questo gene e quindi noi ora andiamo a fare uno zoom su questi enhancer per capirci qualcosa di più.
Vi ricordo che le barrette rosso/viola intenso indicano che il segnale è fuori scala, quindi se io voglio veramente capire tutto questo segnale cosa è posso andarmi a cambiare la scala. Posso andare a configurare il H3K27Ac dicendogli che il massimo invece di 100 potrebbe essere 500 per esempio, così abbasso tutto l’eventuale background e continuo a vedere i picchi che risultano particolarmente elevati. Quindi dopo aver fatto “zoom in” vogliamo andare a vedere se i picchi, cambiando scala siano meglio visualizzabili. Cliccando con il tasto destro su H3K27Ac vado su “Configure Layered H3K27Ac” e possiamo cambiare la scala. Dopo il click trovo questa schermata:
Io avevo impostato su 500 e si erano un po’ troppo appiattiti i picchi quindi proviamo a farlo a 300, quindi sto solo variando la scala per rendermi conto di quali tra i picchi più alti siano i più interessanti. E stiamo guardando non il H3K4me1, che a questo punto nascondo. Vado in “vertical viewing range” e alla voce “max” cambio il valore da 100, settato di base, a 300, poi “submit”. In più per nascondere il picco di H3K4me1 : premo tasto destro in corrispondenza della linea di H3K4me1 sul grafico e imposto “hide” (subito viene nascosto). Questo è il risultato: su H3K27ac non si vedono più le barrette rosse e picchi sono più risolti. La scala ovviamente non posso deciderla a priori ma la imposto in base alla regione, quindi non andrà sempre bene 300 o 100 come impostazione per ogni gene! Se vedo che è fuori scala provo ad abbassarla. Abbasso la scala perché questo mi permette di evidenziare i picchi più alti che sono quelli di maggiore interesse. Il puntino rosso sui picchi di H3K4Me3 mi dice che lì è ancora fuori scala perché sono più alti, in realtà la scala è da uno a mille mentre loro fanno vedere da uno a cento perché già è indicativo sperimentalmente; sperimentalmente vuol dire che c’è moltissimo H3K27 acetilato. Proprio queste zone con i “Puntini rossi” sono interessanti perché il segnale è molto forte , quindi più elevati sono i picchi più sono di interesse.
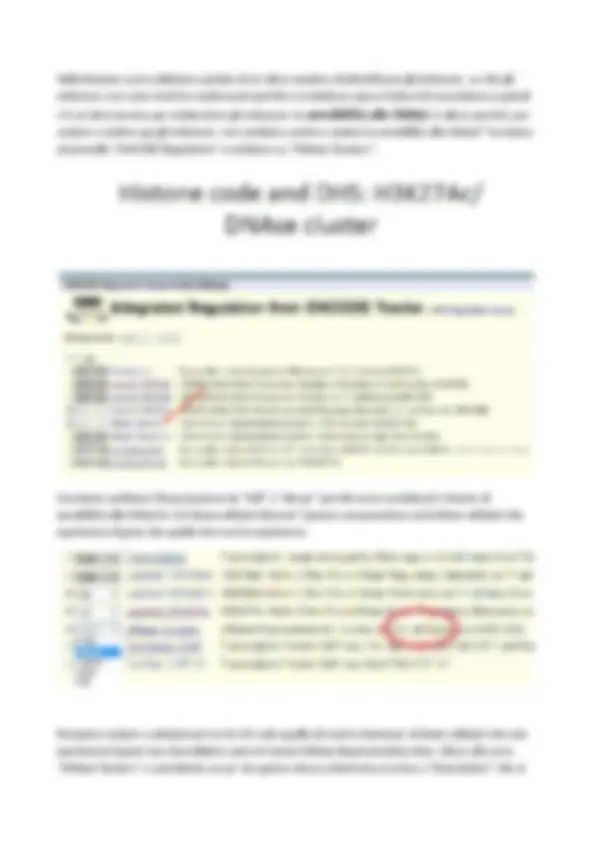
consiglio sempre di leggere perché vi dice che cosa è che state guardando, anche durante l’esame se avete dubbi leggetela così capite cosa state facendo. Quindi questo non è altro che il sommario delle 125 linee cellulari, se a me interessa guardare la mia linea cellulare di interesse posso andarmela a selezionare alla voce “Uniform DNAseI HS”: ( N.B. dal link in “Methods” e non in “Descriptions”!) A cosa serve? Immaginiamo che il mio gene sia regolato in dieci tessuti e sospetto che sia regolato in maniera diversa in ogni tessuto, quindi posso andare a vedere in ogni singolo tessuto dove si trovano i DNAse Hypersensitive sites. In questo caso, per esempio, potremmo non solo prendere le NHEK, che sicuramente lo esprimono, ma anche cercarci qualche altra linea cellulare che lo esprime. Esempio le Human Mammary epithelial Cells (HMEC),( non bisogna saperle a memoria ovviamente, basta controllare) e possiamo mettere le Urothelia. Cliccando sul link indicato dalla freccia si apre la schermata della slide seguente in cui la prof ha deselezionato le linee cellulari impostate di default e ha lasciato solo NHEK, in più, in classe, è andata a selezionare le HMEC e le Urothelia dalla lunga lista dei 125 tipi cellulari presenti, quindi stiamo andando a vedere i siti sensibili alle DNAsi in 3 diverse linee cellulari che io so esprimono il gene P63.
Premo “submit” in fondo a tutto per dare il comando e ottengo questo sulla schermata principale: I puntini colorati indicano Hmec DNAse (il primo), Nhec DNAse (il secondo), Urothelia DNAse (il terzo). Comunque è sempre riportato a margine a sx della schermata a quale linea cellulare si riferiscono i dati.
Vorrei farvi notare che il sito di ipersensibilità delle DNAsi si trova a cavallo dei siti degli istoni sia metilati che acetilati. Ve lo aspettate questo o lo trovate strano? Nei due picchi di H3K27ac sappiamo che ci sono un enhancer per ciascun sito, ma so che questi istoni si trovano sul nucleosoma, e i nucleosomi devono fare un po’ di spazio per far entrare i fattori di trascrizione. Quindi questo avallamento in realtà corrisponde a dove si siedono i fattori di trascrizione e quindi dove si trova il picco di DNAse Hypersensitivity site. Quindi i fattori di trascrizione si trovano tra i due nucleosomi che si sono un po’ distanziati e qui sono seduti la maggior parte dei fattori di trascrizione. Adesso vi convincerò di questo mostrandovi una cosa che però posso solo farvi vedere e che voi non potete riprodurre sui vostri pc, quindi dovete seguirmi. C’è la possibilità di guardare questa cosa con i fattori di trascrizione, con delle Chip-seq per fattori di trascrizione, si può fare sia, come vedremo, su Genome Browser, sia aggiungere il cosidetto “custom track” su Genome Browser. Faccio l’esperimento di Chip-seq con il mio fattore di trascrizione e aggiungo tutti i dati su Genome Browser e me li tengo per me o per chiunque altro voglia vederli. Questa è una cosa che noi abbiamo ma che voi non avete sul pc, vado esattamente nella stessa regione in cui siamo adesso e vi faccio vedere effettivamente dove c’è P63. Uso la versione 2006, leggermente diversa rispetto al vostro che è 2009, risulta leggermente diverso ma non è questo il motivo per cui non potete vedere tutto, semplicemente non avete la possibilità di inserire voi i dati
su Genome Browser. La mia regione di interesse è il grosso introne in cui vedo i grossi picchi di H3K27ac. In corrispondenza di questi picchi io ho i miei picchi “custom track” ottenuti da una Chip-seq con il mio fattore di trascrizione di interesse, in questo caso P63 stesso ( picchi azzurri pag. seguente ). Vi ricordo, ve lo ho spiegato, che P63 ha una autoregolazione su se stesso. Quindi il fattori di trascrizione P63 si lega sul genoma all’interno del gene P63 per autoregolare se stesso. Se io prendo uno di questi due picchi ( i più alti nella zona cerchiata ) e mi avvicino moltissimo vedo un picco di binding di P63, un avallamento tra i due istoni H3K27ac, quindi manca il nucleosoma in quella parte centrale, è leggermente aperto, a questo corrisponde un cluster di DNAse Hypersensity sites. Inoltre qui, come vedremo settimana prossima, c’è una altissima conservazione in mammiferi. Questa regione è un’enhancer, ci si siede un fattore di trascrizione, almeno uno, o molti di più. Noi abbiamo dimostrato sperimentalmente che questo, (P63), si siede in questa regione, che c’è acetilazione degli istoni H3K27, non c’è trimetilazione di H3K4 perché ovviamente quello è presente solo nei promotori, di questo c’è un piccolo picco ma non è molto significativo, e poi c’è un DNAse Hypersensitivity site. Ma ora mi chiedo: come faccio ad sapere quale è il consensus di legame al DNA? D: Quanto grande è il consensus dei fattori di trascrizione? È molto piccolo, tra 4 e 20 bp. slide non mostrata a lezione. N° 21/32 file “lezione 3 bioinformatica”. Se io vado a stringere sempre di più tenendo in considerazione sia il picco che il sito di DNAse Hypersensitivity ad un certo punto se stringo sufficientemente vedo un pezzettino così piccolo che posso andarmi a prendere in “view” la